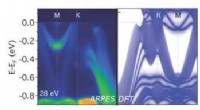
Los nuevos modelos de aprendizaje profundo predicen las interacciones entre átomos en moléculas orgánicas. Estos modelos ayudarán a los biólogos computacionales y a los investigadores en desarrollo de fármacos a comprender y tratar las enfermedades. Crédito:Laboratorio Nacional de Los Alamos
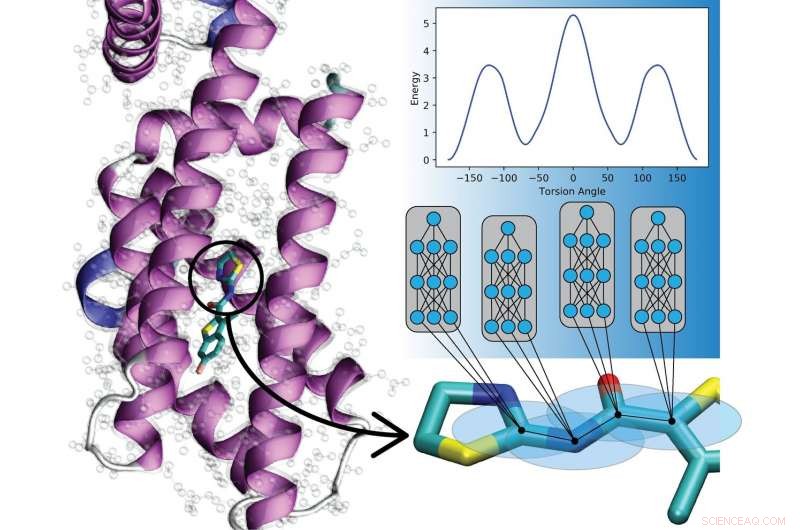
Nuevo trabajo del Laboratorio Nacional de Los Alamos, la Universidad de Carolina del Norte en Chapel Hill, y la Universidad de Florida está demostrando que las redes neuronales artificiales se pueden entrenar para codificar las leyes de la mecánica cuántica para describir los movimientos de las moléculas, simulaciones de sobrealimentación potencialmente en una amplia gama de campos.
"Esto significa que ahora podemos modelar materiales y dinámica molecular miles de millones de veces más rápido en comparación con los métodos cuánticos convencionales". manteniendo el mismo nivel de precisión, "dijo Justin Smith, Físico de Los Alamos y miembro de Metropolis en la División Teórica del laboratorio. Comprender cómo se mueven las moléculas es fundamental para aprovechar su valor potencial para el desarrollo de fármacos, simulaciones de proteínas y química reactiva, por ejemplo, y tanto la mecánica cuántica como los métodos experimentales (empíricos) se incorporan a las simulaciones.
La nueva técnica, llamado potencial ANI-1ccx, promete promover las capacidades de los investigadores en muchos campos y mejorar la precisión de los potenciales basados en el aprendizaje automático en futuros estudios de aleaciones de metales y física de detonaciones.
Algoritmos de mecánica cuántica (QM), utilizado en computadoras clásicas, Puede describir con precisión los movimientos mecánicos de un compuesto en su entorno operativo. Pero QM escala muy mal con diferentes tamaños moleculares, limitando severamente el alcance de posibles simulaciones. Incluso un ligero aumento en el tamaño molecular dentro de una simulación puede aumentar drásticamente la carga computacional. Por eso, los profesionales suelen recurrir al uso de información empírica, que describe el movimiento de los átomos en términos de la física clásica y las leyes de Newton, permitiendo simulaciones que escalan a miles de millones de átomos o millones de compuestos químicos.
Tradicionalmente, Los potenciales empíricos han tenido que encontrar un compromiso entre precisión y transferibilidad. Cuando los muchos parámetros del potencial están finamente ajustados para un compuesto, la precisión disminuye en otros compuestos.
En lugar de, el equipo de Los Alamos, con la Universidad de Carolina del Norte en Chapel Hill y la Universidad de Florida, ha desarrollado un enfoque de aprendizaje automático llamado aprendizaje de transferencia que les permite desarrollar potenciales empíricos aprendiendo de los datos recopilados sobre millones de otros compuestos. El nuevo enfoque con el potencial empírico del aprendizaje automático se puede aplicar a nuevas moléculas en milisegundos, permitiendo la investigación de un número mucho mayor de compuestos en escalas de tiempo mucho más largas.