
Shantenu Jha, presidente del Centro de Descubrimiento Basado en Datos de Brookhaven Lab, y su equipo de la Universidad de Rutgers y el University College de Londres diseñaron un marco de software para calcular con precisión y rapidez la fuerza con la que los candidatos a fármacos se unen a sus proteínas objetivo. El marco tiene como objetivo resolver el problema del mundo real del diseño de fármacos, que actualmente es un proceso largo y costoso, y podría tener un impacto en la medicina personalizada. Crédito:Laboratorio Nacional Brookhaven
Las soluciones a muchos problemas científicos y de ingeniería del mundo real, desde la mejora de los modelos meteorológicos y el diseño de nuevos materiales energéticos hasta la comprensión de cómo se formó el universo, requieren aplicaciones que puedan escalar a un tamaño muy grande y un alto rendimiento. Cada año, a través de su International Scalable Computing Challenge (SCALE), el Instituto de Ingenieros Eléctricos y Electrónicos (IEEE) reconoce un proyecto que promueve el desarrollo de aplicaciones y la infraestructura de soporte para permitir el desarrollo a gran escala, la informática de alto rendimiento necesaria para resolver estos problemas.
El ganador de este año, "Habilitación de la compensación entre precisión y costo computacional:algoritmos adaptables para reducir el tiempo de comprensión clínica, "es el resultado de una colaboración entre químicos y científicos de computación y computación en el Laboratorio Nacional Brookhaven del Departamento de Energía de EE. UU. (DOE), Universidad Rutgers, y University College London. Los miembros del equipo fueron honrados en el 18o Simposio Internacional de Clúster de IEEE / Association for Computing Machinery (ACM), Computación en la nube y en red celebrada en Washington, CORRIENTE CONTINUA, del 1 al 4 de mayo.
"Desarrollamos una metodología de cálculo numérico para evaluar con precisión y rapidez la eficacia de diferentes fármacos candidatos, "dijo el miembro del equipo Shantenu Jha, presidente del Center for Data-Driven Discovery, parte de la Iniciativa de Ciencias Computacionales de Brookhaven Lab. "Aunque todavía no hemos aplicado esta metodología para diseñar un nuevo fármaco, demostramos que podría funcionar a gran escala involucrada en el proceso de descubrimiento de fármacos ".
El descubrimiento de fármacos es como diseñar una llave que se ajuste a una cerradura. Para que un medicamento sea eficaz en el tratamiento de una enfermedad en particular, debe unirse estrechamente a una molécula, generalmente una proteína, que está asociada con esa enfermedad. Solo entonces el fármaco puede activar o inhibir la función de la molécula diana. Los investigadores pueden evaluar 10, 000 o más compuestos moleculares antes de encontrar alguno que tenga la actividad biológica deseada. Pero estos compuestos de "plomo" a menudo carecen de la potencia, selectividad, o estabilidad necesaria para convertirse en una droga. Al modificar la estructura química de estos cables, los investigadores pueden diseñar compuestos con las propiedades similares a las de las drogas adecuadas. Los candidatos a fármacos diseñados luego se mueven a lo largo del proceso de desarrollo hasta la etapa de prueba preclínica. De estos candidatos, solo una pequeña fracción entra en la fase de ensayo clínico, y solo uno termina convirtiéndose en un medicamento aprobado para el uso del paciente. Llevar un nuevo medicamento al mercado puede llevar una década o más y costar miles de millones de dólares.
Superar los cuellos de botella del diseño de fármacos a través de la ciencia computacional
Los avances recientes en tecnología y conocimiento han dado lugar a una nueva era de descubrimiento de fármacos, que podría reducir significativamente el tiempo y los gastos del proceso de desarrollo de fármacos. Las mejoras en nuestra comprensión de las estructuras cristalinas tridimensionales de las moléculas biológicas y los aumentos en la potencia de cálculo están haciendo posible el uso de métodos computacionales para predecir las interacciones fármaco-objetivo.
El descubrimiento de fármacos es un problema de candado y llave en el que el fármaco (llave) debe ajustarse específicamente al objetivo biológico (candado). Crédito:Laboratorio Nacional Brookhaven
En particular, una técnica de simulación por computadora llamada dinámica molecular ha demostrado ser prometedora para predecir con precisión la fuerza con la que las moléculas del fármaco se unen a sus objetivos (afinidad de unión). La dinámica molecular simula cómo se mueven los átomos y las moléculas a medida que interactúan en su entorno. En el caso del descubrimiento de fármacos, las simulaciones revelan cómo las moléculas del fármaco interactúan con su proteína objetivo y cambian la conformación de la proteína, o forma, que determina su función.
Sin embargo, estas capacidades de predicción aún no están operando a una escala lo suficientemente grande ni a una velocidad lo suficientemente rápida como para que las compañías farmacéuticas las adopten en su proceso de desarrollo.
"Traducir estos avances en precisión predictiva para impactar la toma de decisiones industriales requiere que del orden de 10, 000 afinidades de unión se calculan lo más rápido posible, sin la pérdida de precisión, ", dijo Jha." La producción de información oportuna exige una eficiencia computacional que se basa en el desarrollo de nuevos algoritmos y sistemas de software escalables, y la asignación inteligente de recursos de supercomputación ".
Jha y sus colaboradores en la Universidad de Rutgers, donde también es profesor en el Departamento de Ingeniería Eléctrica e Informática, y el University College London diseñaron un marco de software para respaldar el cálculo rápido y preciso de las afinidades de enlace mientras se optimiza el uso de los recursos computacionales. Este marco, denominada Calculadora de afinidad de enlace de alto rendimiento (HTBAC), se basa en el proyecto RADICAL-Cybertools que Jha lidera como investigador principal del Laboratorio de Investigación de Rutgers en Ciberinfraestructura y Aplicaciones Distribuidas Avanzadas (RADICAL). El objetivo de RADICAL-Cybertools es proporcionar un conjunto de componentes básicos de software para respaldar los flujos de trabajo de aplicaciones científicas a gran escala en plataformas informáticas de alto rendimiento. que agregan poder de computación para resolver grandes problemas computacionales que de otra manera serían irresolubles debido al tiempo requerido.
En informática, Los flujos de trabajo se refieren a una serie de pasos de procesamiento necesarios para completar una tarea o resolver un problema. Especialmente para flujos de trabajo científicos, Es importante que los flujos de trabajo sean flexibles para que puedan adaptarse dinámicamente durante el tiempo de ejecución para proporcionar los resultados más precisos mientras se hace un uso eficiente del tiempo de computación disponible. Estos flujos de trabajo adaptativos son ideales para el descubrimiento de fármacos porque solo deben evaluarse más a fondo los fármacos con altas afinidades de unión.
"La compensación deseada entre la precisión requerida y el costo (tiempo) computacional cambia a lo largo del descubrimiento del fármaco a medida que el proceso pasa de la detección a la selección de prospectos y luego a la optimización de prospectos, ", dijo Jha." Un número significativo de compuestos deben ser analizados a bajo costo para eliminar los aglutinantes deficientes antes de que se necesiten métodos más precisos para discriminar los mejores aglutinantes. Proporcionar el tiempo de solución más rápido requiere monitorear el progreso de las simulaciones y basar las decisiones sobre la ejecución continua en la importancia científica ".
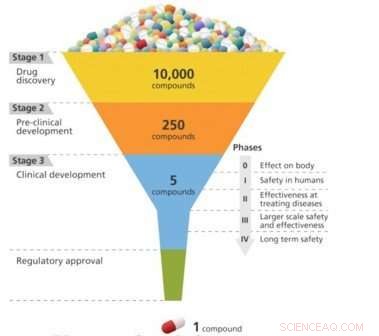
Un esquema del proceso de desarrollo de fármacos, que se concentra progresivamente en los candidatos más eficaces de un gran grupo inicial. Crédito:Laboratorio Nacional Brookhaven
En otras palabras, No tendría sentido continuar con las simulaciones de una interacción fármaco-proteína en particular si el fármaco se une débilmente a la proteína en comparación con los otros candidatos. Pero tendría sentido asignar recursos computacionales adicionales si un fármaco muestra una alta afinidad de unión.
Apoyar los flujos de trabajo adaptativos a gran escala, característicos de los programas de descubrimiento de fármacos, requiere capacidades computacionales avanzadas. HTBAC proporciona dicho soporte a través de una capa de software de middleware flexible que permite la ejecución adaptativa de algoritmos. En la actualidad, HTBAC admite dos algoritmos:muestreo mejorado de dinámica molecular con aproximación de disolvente continuo (ESMACS) e integración termodinámica con muestreo mejorado (TIES). ESMACS, un método computacionalmente más barato pero menos riguroso que TIES, calcula la fuerza de unión de un fármaco a su proteína diana sobre la base de simulaciones de dinámica molecular. Por el contrario, TIES compara las afinidades de unión relativas de dos fármacos diferentes con la misma proteína.
"ESMACS proporciona un enfoque cuantitativo rápido lo suficientemente sensible para determinar las afinidades de unión para que podamos eliminar los aglutinantes deficientes, mientras que TIES proporciona un método más preciso para investigar buenos aglutinantes a medida que se refinan y mejoran, "dijo Jumana Dakka, un doctorado de segundo año. estudiante de Rutgers y miembro del grupo RADICAL.
Para determinar qué algoritmo ejecutar, HTBAC analiza los cálculos de afinidad de unión en tiempo de ejecución. Este análisis informa las decisiones sobre el número de simulaciones simultáneas a realizar y si se deben agregar o eliminar pasos de estimulación para cada fármaco candidato investigado.
Poniendo el marco a prueba
Jha's team demonstrated how HTBAC could provide insight from drug candidate data on a short timescale by reproducing results from a collaborative study between University College London and the London-based pharmaceutical company GlaxoSmithKline to discover drug compounds that bind to the BRD4 protein. Known to play a key role in driving cancer and inflammatory diseases, the BRD4 protein is a major target of bromodomain-containing (BRD) inhibitors, a class of pharmaceutical drugs currently being evaluated in clinical trials. The researchers involved in this collaborative study are focusing on identifying promising new drugs to treat breast cancer while developing an understanding of why certain drugs fail in the presence of breast cancer gene mutations.
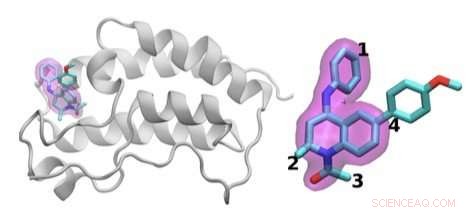
The scientists investigated the chemical structures of 16 drugs based on the same tetrahydroquinoline (THQ) scaffold. On the left is a cartoon of the BRD4 protein bound to one of these drugs; on the right is a molecular representation of a drug with the THQ scaffold highlighted in magenta. Regions that are chemically modified between the drugs investigated in this study are labeled 1 to 4. Typically, only a small change is made to the chemical structure of one drug to the next. This conservative approach makes it easier for researchers to understand why one drug is effective, whereas another is not. Crédito:Laboratorio Nacional Brookhaven
Jha and his team concurrently screened a group of 16 closely related drug candidates from the study by running thousands of computational sequences on more than 32, 000 computing cores. They ran the computations on the Blue Waters supercomputer at the National Center for Computing Applications, University of Illinois at Urbana-Champaign.
In a real drug design scenario, many more compounds with a wider range of chemical properties would need to be investigated. The team members previously demonstrated that the workload management layer and runtime system underlying HTBAC could scale to handle 10, 000 concurrent tasks.
"HTBAC could support the concurrent screening of different compounds at unprecedented scales—both in the number of compounds and computational resources used, " said Jha. "We showed that HTBAC has the ability to solve a large number of drug candidates in essentially the same amount of time it would take to solve a smaller set, assuming the number of processors increases proportionally with the number of candidates."
This ability is made possible through HTBAC's adaptive functionality, which allows it to execute the optimal algorithm depending on the properties of the drugs being investigated, improving the accuracy of the results and minimizing compute time.
"The lead optimization stage usually considers on the order of 10, 000 small molecules, " said Jha. "While experiment automation reduces the amount of time needed to calculate the binding affinities, HTBAC has the potential to cut this time (and cost) by an order of magnitude or more."
With HTBAC, TIES requires approximately 25, 000 central processing unit (CPU) core hours for a single prediction. At least a 250 million core hours would be needed for a large-scale study to support a pharmaceutical drug screening campaign, with a typical turnaround time of about two weeks. HTBAC could facilitate running studies requiring sustained usage of millions of core hours per day.
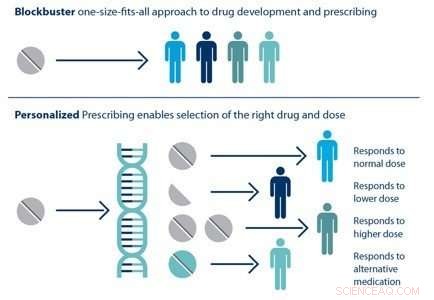
Individual patients respond differently to drugs. The ability to predict which treatment is best for a particular patient based on his or her genetic sequence is the goal of personalized medicine. Crédito:Laboratorio Nacional Brookhaven
When the University of College London–GlaxoSmithKline study concludes, Jha and his team hope to be given the experimental data on the tens of thousands of drug candidates, without knowing which candidate ended up being the best one. Con esta información, they could perform a blind test to determine whether HTBAC provides an improvement in compute time (for a given accuracy) over the existing automated methods for drug discovery. Si necesario, they could then refine their methodology.
Applying scalable computing to precision medicine
HTBAC not only has the potential to improve the speed and accuracy of drug discovery in the pharmaceutical industry but also to improve individual patient outcomes in clinical settings. Using target proteins based on a patient's genetic sequence, HTBAC could predict a patient's response to different drug treatments. This personalized assessment could replace the traditional one-size-fits-all approach to medicine. Por ejemplo, such predictions could help determine which cancer patients would actually benefit from chemotherapy, avoiding unnecessary toxicity.
According to Jha, the computation time would have to be significantly reduced in order for physicians to clinically use HTBAC to treat their patients:"Our grand vision is to apply scalable computing techniques to personalized medicine. If we can use these techniques to optimize drugs and drug cocktails for each individual's unique genetic makeup on the order of a few days, we will be empowered to treat diseases much more effectively."
"Extreme-scale computing for precision medicine is an emerging area that CSI and Brookhaven at large have begun to tackle, " said CSI Director Kerstin Kleese van Dam. "This work is a great example of how technologies we originally developed to tackle DOE challenges can be applied to other domains of high national impact. We look forward to forming more strategic partnerships with other universities, pharmaceutical companies, and medical institutions in this important area that will transform the future of health care."














