Los investigadores han desarrollado una plataforma que combina experimentos automatizados con IA para predecir cómo reaccionarán los químicos entre sí, lo que podría acelerar el proceso de diseño de nuevos medicamentos.
Predecir cómo reaccionarán las moléculas es vital para el descubrimiento y la fabricación de nuevos productos farmacéuticos, pero históricamente esto ha sido un proceso de prueba y error, y las reacciones a menudo fallan. Para predecir cómo reaccionarán las moléculas, los químicos suelen simular electrones y átomos en modelos simplificados, un proceso computacionalmente costoso y a menudo inexacto.
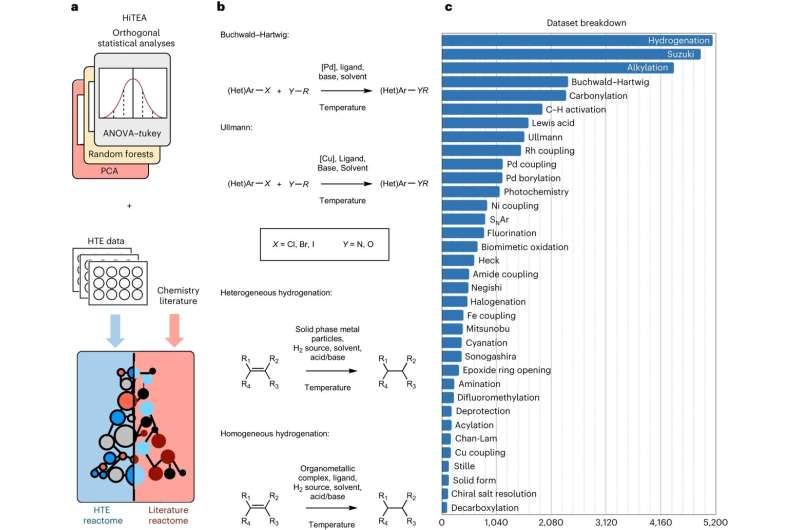
Ahora, investigadores de la Universidad de Cambridge han desarrollado un enfoque basado en datos, inspirado en la genómica, en el que se combinan experimentos automatizados con aprendizaje automático para comprender la reactividad química, lo que acelera enormemente el proceso. Llamaron a su enfoque, que fue validado en un conjunto de datos de más de 39.000 reacciones farmacéuticamente relevantes, el "reactoma" químico.
Sus resultados, publicados en la revista Nature Chemistry , son producto de una colaboración entre Cambridge y Pfizer.
"El reactor podría cambiar la forma en que pensamos sobre la química orgánica", dijo la Dra. Emma King-Smith del Laboratorio Cavendish de Cambridge, primera autora del artículo. "Una comprensión más profunda de la química podría permitirnos fabricar productos farmacéuticos y muchos otros productos útiles mucho más rápido. Pero lo más fundamental es que la comprensión que esperamos generar será beneficiosa para cualquiera que trabaje con moléculas".
El enfoque del reactor selecciona correlaciones relevantes entre reactivos, reactivos y el rendimiento de la reacción a partir de los datos, y señala lagunas en los datos mismos. Los datos se generan a partir de experimentos automatizados muy rápidos o de alto rendimiento.
"La química de alto rendimiento ha cambiado las reglas del juego, pero creíamos que había una manera de descubrir una comprensión más profunda de las reacciones químicas que la que se puede observar a partir de los resultados iniciales de un experimento de alto rendimiento", dijo King-Smith.>
"Nuestro enfoque descubre las relaciones ocultas entre los componentes de la reacción y los resultados", afirmó el Dr. Alpha Lee, quien dirigió la investigación. "El conjunto de datos con el que entrenamos el modelo es enorme:ayudará a que el proceso de descubrimiento químico pase del método de prueba y error a la era del big data".
En un artículo relacionado, publicado en Nature Communications , el equipo desarrolló un enfoque de aprendizaje automático que permite a los químicos introducir transformaciones precisas en regiones moleculares preespecificadas, lo que permite un diseño de fármacos más rápido.
El enfoque permite a los químicos modificar moléculas complejas (como un cambio de diseño de último momento) sin tener que crearlas desde cero. Fabricar una molécula en el laboratorio suele ser un proceso de varios pasos, como construir una casa. Si los químicos quieren variar el núcleo de una molécula, la forma convencional es reconstruir la molécula, como derribar una casa y reconstruirla desde cero. Sin embargo, las variaciones centrales son importantes para el diseño de medicamentos.
Una clase de reacciones conocidas como reacciones de funcionalización de etapa tardía intenta introducir transformaciones químicas directamente en el núcleo, evitando la necesidad de comenzar desde cero. Sin embargo, es un desafío hacer que la funcionalización en la última etapa sea selectiva y controlada; generalmente hay muchas regiones de las moléculas que pueden reaccionar y es difícil predecir el resultado.
"Las funcionalizaciones de última etapa pueden producir resultados impredecibles y los métodos actuales de modelado, incluida nuestra propia intuición experta, no son perfectos", dijo King-Smith. "Un modelo más predictivo nos daría la oportunidad de realizar una mejor detección".
Los investigadores desarrollaron un modelo de aprendizaje automático que predice dónde reaccionaría una molécula y cómo varía el sitio de reacción en función de las diferentes condiciones de reacción. Esto permite a los químicos encontrar formas de modificar con precisión el núcleo de una molécula.
"Preentrenamos el modelo con una gran cantidad de datos espectroscópicos (enseñando efectivamente al modelo química general) antes de ajustarlo para predecir estas intrincadas transformaciones", dijo King-Smith. Este enfoque permitió al equipo superar la limitación de la escasez de datos:hay relativamente pocas reacciones de funcionalización en etapa tardía reportadas en la literatura científica. El equipo validó experimentalmente el modelo en un conjunto diverso de moléculas similares a fármacos y pudo predecir con precisión los sitios de reactividad en diferentes condiciones.
"La aplicación del aprendizaje automático a la química a menudo se ve frenada por el problema de que la cantidad de datos es pequeña en comparación con la inmensidad del espacio químico", dijo Lee. "Nuestro enfoque (diseñar modelos que aprenden de grandes conjuntos de datos que son similares pero no iguales al problema que intentamos resolver) resuelve este desafío fundamental de la escasez de datos y podría desbloquear avances más allá de la funcionalización en la última etapa".
Más información: Emma King-Smith et al, Sondeando el 'reactoma' químico con datos de experimentación de alto rendimiento, Nature Chemistry (2024). DOI:10.1038/s41557-023-01393-w
Funcionalización de etapa tardía de Predictive Minisci con aprendizaje por transferencia, Nature Communications (2024). DOI:10.1038/s41467-023-42145-1. www.nature.com/articles/s41467-023-42145-1
Información de la revista: Comunicaciones sobre la naturaleza , Química de la Naturaleza
Proporcionado por la Universidad de Cambridge