Un equipo de investigación de Skoltech presentó un nuevo método que aprovecha el aprendizaje automático para estudiar las propiedades de policristales, compuestos y sistemas multifásicos. Logró una alta precisión, casi tan buena como la de los métodos mecánico-cuánticos, que sólo son aplicables a materiales con menos de unos pocos cientos de átomos.
El nuevo método también se beneficia del aprendizaje activo sobre entornos atómicos locales. El artículo está publicado en Teoría y simulaciones avanzadas. diario.
"Muchos materiales industriales se sintetizan como policristales o sistemas multifásicos. Contienen tanto un monocristal como componentes amorfos entre los granos monocristalinos. La gran cantidad de átomos dificulta el cálculo de las propiedades de estos sistemas utilizando métodos modernos de mecánica cuántica. Densidad funcional La teoría sólo se puede aplicar a materiales con unos pocos cientos de átomos."
"Para abordar el problema utilizamos un enfoque de aprendizaje automático basado en los potenciales momento tensoriales (MTP). Estos potenciales también se han desarrollado en Skoltech bajo la dirección del profesor Alexander Shapeev", comentó Faridun Jalolov, autor principal del estudio y un doctorado en Skoltech. estudiante del programa de Ciencia e Ingeniería de Materiales.
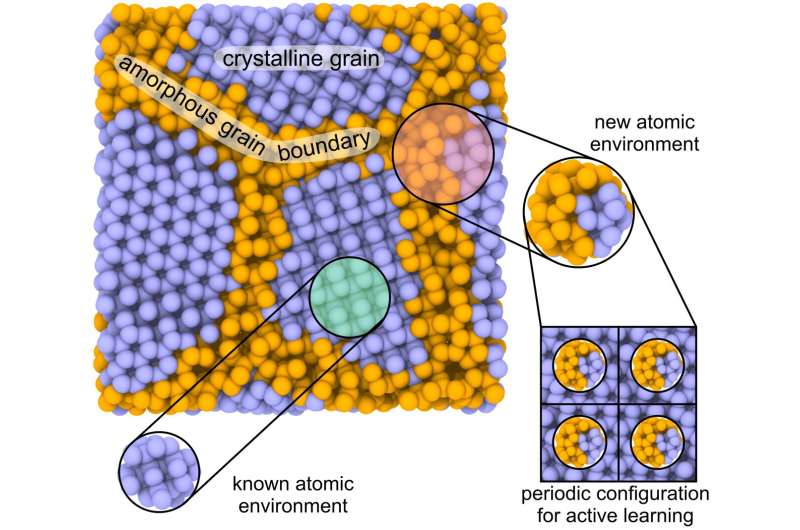
En comparación con otras soluciones, los autores ven el potencial del nuevo método en el aprendizaje activo en entornos atómicos locales. Al calcular una estructura grande con muchos cientos de miles de átomos, el MTP identifica qué átomo comete un error en los cálculos o se calcula incorrectamente. La razón de esto podría ser el conjunto de datos de entrenamiento limitado, que impide considerar todas las configuraciones posibles del sistema.
A continuación se "corta" el entorno local de este átomo y se calcula su energía mediante la mecánica cuántica. Luego, los datos se vuelven a agregar al conjunto de entrenamiento para seguir aprendiendo. A medida que avanza el aprendizaje sobre la marcha, los cálculos continúan hasta que encuentran otra configuración que debe incluirse en el proceso de capacitación. Otros potenciales conocidos del aprendizaje automático no se pueden aprender en pequeñas partes locales de estructuras grandes, lo que limita su aplicabilidad y precisión.
"A modo de ejemplo, estudiamos las propiedades mecánicas de los policristales de diamante, que son los materiales naturales más duros y se utilizan a menudo en la industria, por ejemplo, en la fabricación de equipos de perforación para pozos petroleros. Los resultados muestran que las propiedades mecánicas de estos diamantes policristalinos dependen depende del tamaño del grano:cuanto más grande es el grano, más similares son las propiedades a las de un diamante monocristalino", continuó Jalolov.
Los autores señalaron que este enfoque permitirá estudiar las propiedades mecánicas de materiales no monocristalinos que normalmente se sintetizan y utilizan en experimentos, así como realizar estudios integrales de materiales policristalinos y compuestos y obtener datos lo más cercanos posible a los resultados experimentales. /P>
"En el uso real, con frecuencia se emplean materiales que no son cristales perfectos debido a su incapacidad para que los cristales perfectos cumplan completamente con los requisitos de una pieza específica de equipo".
"Un buen ejemplo de esto es el carburo de tungsteno y el cobalto. Al agregar cobalto al carburo de tungsteno, el material se vuelve más resistente a las grietas, lo que lo hace muy valioso en las aplicaciones. El nuevo método nos permitirá investigar las causas y formas de alterar la mecánica propiedades de estos sistemas multifásicos a nivel atómico", afirmó Alexander Kvashnin, director de la investigación y profesor del Centro de Transición Energética.
Más información: Faridun N. Jalolov et al, Propiedades mecánicas de sólidos simples y policristalinos a partir del aprendizaje automático, Teoría y simulaciones avanzadas (2024). DOI:10.1002/adts.202301171
Proporcionado por el Instituto de Ciencia y Tecnología de Skolkovo