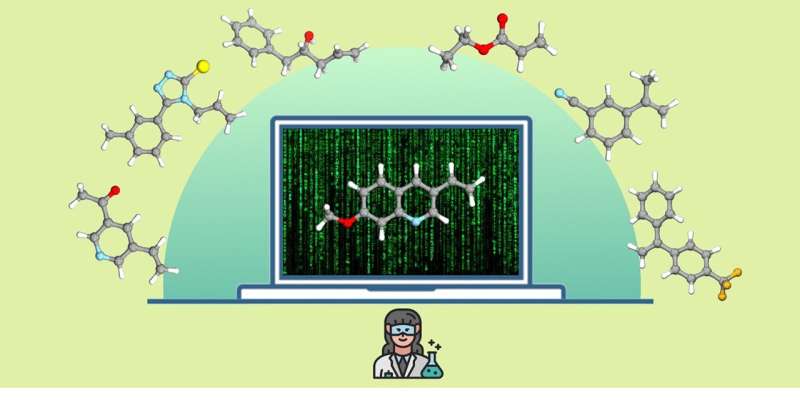
Los químicos suelen desarrollar y optimizar nuevas reacciones químicas utilizando los llamados sistemas modelo, es decir, sustratos simples y de fácil acceso. Luego utilizan hasta alrededor de 100 sustratos más como ejemplos para demostrar que la reacción funciona. Esta demostración de aplicabilidad versátil se denomina "alcance" en la jerga técnica.
Sin embargo, una selección subjetiva de los sustratos a menudo da como resultado una imagen distorsionada de las posibilidades de aplicación de la reacción recientemente desarrollada. A menudo no está claro si se puede utilizar para sintetizar el producto deseado. Para abordar este problema, un equipo dirigido por el químico profesor Frank Glorius de la Universidad de Münster (Alemania) propone un método libre de sesgos, asistido por ordenador, para seleccionar los sustratos modelo para evaluar nuevas reacciones químicas.
La selección de sustratos se basa en la complejidad y las propiedades estructurales de compuestos farmacéuticos reales. "Nuestro método tiene como objetivo mejorar la calidad y el contenido informativo de los datos de reacciones químicas en el futuro y cerrar las lagunas de conocimiento", explica Glorius.
Una comprensión más profunda de las nuevas reacciones reduce las barreras para su aplicación tanto en el contexto académico como industrial. La disponibilidad de datos imparciales y de alta calidad también facilita significativamente el uso del aprendizaje automático y allana el camino para un uso más completo de los datos. El trabajo ha sido publicado en la revista ACS Central Science .
Según los autores del equipo, los intentos de estandarizar y objetivar el desarrollo y la evaluación de reacciones químicas son todavía bastante nuevos y relativamente poco comunes. "Con nuestra publicación nos gustaría iniciar un 'proceso de replanteamiento'. En lugar de realizar tantos experimentos como sea posible, que a menudo están sesgados o tienen un resultado predecible, la atención debería centrarse en obtener los mejores datos posibles sobre nuevas reacciones químicas". dice el primer autor Debanjan Rana.
Otros científicos también han intentado evaluar reacciones químicas basándose en sustratos "mejores" seleccionados. Sin embargo, este trabajo se limitó a casos especiales, ya sea a estructuras firmemente seleccionadas con relevancia farmacéutica o a estructuras especialmente diseñadas para una reacción única, que deben calcularse y seleccionarse en un proceso complejo.
A diferencia del trabajo anterior, el método presentado por el equipo de Münster tiene en cuenta toda la estructura de una molécula, lo que lo hace universalmente aplicable a cualquier reacción química.
Niklas Hölter, uno de los autores del artículo en Münster, explica el proceso de pensamiento detrás del estudio:"El alcance es de importancia central en todas las publicaciones sobre síntesis química. Sin embargo, los químicos a menudo están sesgados en la elección de los compuestos sustrato para probar.
"Por ejemplo, eligen sustratos estructuralmente muy simples, muy similares al sustrato modelo o simplemente disponibles en el laboratorio ('sesgo de selección'). En sus publicaciones, a menudo no mencionan reacciones fallidas para poder pintar. una mejor imagen ('sesgo de información')".
Al sintetizar nuevos compuestos químicos, como ingredientes o materiales activos, los químicos deben seleccionar el método más adecuado para producir el compuesto objetivo a partir de una gran cantidad de reacciones y métodos químicos conocidos. Para ello, consideran varios factores, como el rendimiento del producto deseado, así como aspectos medioambientales y de seguridad. Por lo tanto, el desarrollo de reacciones químicas nuevas y versátiles sigue siendo el foco de la investigación química actual.
El método desarrollado por el equipo de la Universidad de Münster utilizó huellas dactilares moleculares para transferir todos los ingredientes farmacéuticos activos aprobados a un código digital. Utilizando métodos de agrupamiento y aprendizaje automático no supervisado, crearon un modelo que divide este "espacio" de ingredientes farmacéuticos activos en regiones químicamente significativas basadas en estructuras moleculares.
Para evaluar una nueva reacción química, se pueden proyectar miles de posibles sustratos de prueba en el mismo espacio utilizando el modelo de aprendizaje automático. Se selecciona automáticamente un sustrato de prueba desde el centro de cada una de las regiones previamente identificadas para cubrir todo el espacio sin sesgos.
Más información: Debanjan Rana et al, Estandarización de la selección de sustratos:una estrategia hacia una evaluación imparcial de la generalidad de la reacción, ACS Central Science (2024). DOI:10.1021/acscentsci.3c01638
Información de la revista: Ciencia central de ACS
Proporcionado por la Universidad de Münster