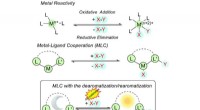
Estructuras generadas y redes de rutas de reacción para los pasos de reacción previstos para la reacción de Strecker (a) y la reacción de Passerini (b). Las flechas blancas muestran la ruta de varios pasos correspondiente al mecanismo de reacción conocido. Crédito:Satoshi Maeda
Los investigadores han superado las limitaciones computacionales para predecir los materiales de partida de las reacciones de varios pasos utilizando solo información sobre la molécula del producto objetivo. Su estudio se publica en JACS Au .
¿Alguna vez solo vio el final de un programa de televisión y se preguntó cómo progresó la historia hasta ese final? De manera similar, los químicos a menudo tienen en mente una molécula deseada y se preguntan qué tipo de reacción podría producirla. Investigadores del Grupo Maeda en el Instituto para el Diseño y Descubrimiento de Reacciones Químicas (ICReDD) y la Universidad de Hokkaido desarrollaron un método que puede predecir la "historia" (es decir, los materiales de partida y las rutas de reacción) de reacciones químicas de varios pasos utilizando solo información sobre el "final" (es decir, las moléculas del producto).
Predecir la receta de la molécula de un producto objetivo, sin más conocimiento que la propia molécula, sería una herramienta poderosa para acelerar el descubrimiento de nuevas reacciones. El grupo de Maeda desarrolló previamente un método computacional que logró predecir reacciones de un solo paso de esta manera. Sin embargo, expandirse a reacciones de múltiples pasos conduce a un aumento dramático en el número de posibles vías de reacción, lo que se conoce como explosión combinatoria. Este fuerte aumento en la complejidad da como resultado costos de cálculo prohibitivamente altos.
Para superar esta limitación, los investigadores desarrollaron un algoritmo que reduce la cantidad de caminos que deben explorarse descartando los caminos menos viables en cada paso de la reacción. Después de calcular todos los caminos posibles para un paso hacia atrás en la reacción, un método de análisis cinético evalúa qué tan bien cada camino produce la molécula objetivo. Las rutas de reacción que no producen la molécula objetivo por encima de un porcentaje de umbral preestablecido no se consideran lo suficientemente significativas y no se exploran más.
Este ciclo de explorar, evaluar y descartar rutas de reacción se repite para cada paso hacia atrás en una reacción de varios pasos y mitiga la explosión combinatoria que ocurriría normalmente, haciendo que las reacciones de varios pasos sean más factibles de calcular. Los métodos anteriores se limitaban a reacciones de un solo paso, mientras que este nuevo método podía predecir reacciones que implicaban más de seis pasos, lo que marca un salto importante en la capacidad.
Como prueba de concepto, los investigadores probaron el método en dos conocidas reacciones de varios pasos, las reacciones de Strecker y Passerini. Se propusieron miles de materiales de partida candidatos para cada reacción, que se filtraron a los candidatos más prometedores en función de la estabilidad y el rendimiento del producto. De manera crítica, entre los candidatos propuestos se encontraban los materiales de partida bien conocidos para cada reacción, lo que confirma la capacidad de la técnica para identificar materiales de partida experimentalmente viables solo a partir de la molécula del producto objetivo.
Aunque se requiere más trabajo para permitir la predicción de sistemas aún más grandes y complejos, los investigadores anticipan que este avance en el manejo de procesos de varios pasos acelerará el descubrimiento de nuevas reacciones químicas.
"Este trabajo proporciona un enfoque único, ya que es la primera vez que es posible realizar predicciones inversas de reacciones de varios pasos utilizando cálculos químicos cuánticos sin utilizar ningún conocimiento o datos sobre la reacción", dijo el profesor Satoshi Maeda. "Esperamos que esta técnica permita el descubrimiento de transformaciones químicas completamente inimaginables, en cuyo caso hay poco conocimiento o datos experimentales para usar". El nuevo método de síntesis química asistido por computadora reduce el tiempo y el costo de la investigación