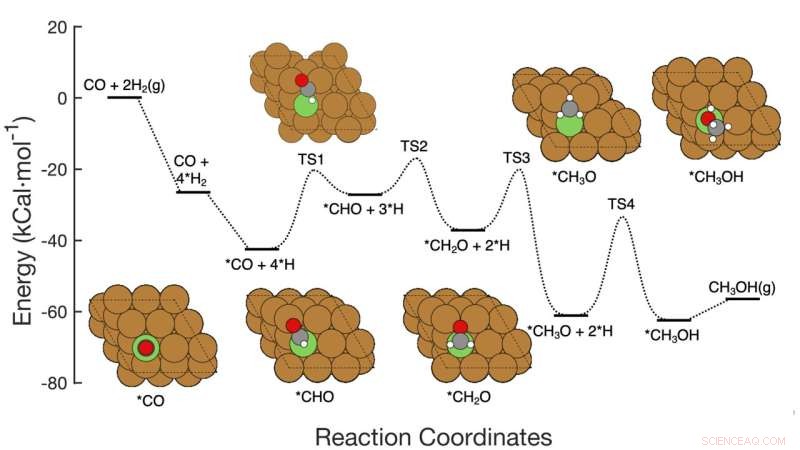
Este gráfico muestra la ruta de reacción de siete pasos de la hidrogenación del CO2 a metanol sobre catalizadores a base de cobre, incluidos los reactivos en cada paso, los arreglos atómicos esquemáticos de los intermedios y las barreras de activación de energía necesarias para pasar de un paso a otro. El equipo de Brookhaven Lab demostró un marco de aprendizaje automático que identificó con éxito qué pasos/combinaciones de pasos modificar para mejorar la producción de metanol. Su trabajo podría ayudar a guiar el diseño de nuevos catalizadores para lograr ese objetivo y el marco se puede aplicar para optimizar otras reacciones. Crédito:Laboratorio Nacional de Brookhaven
Los químicos del Laboratorio Nacional Brookhaven del Departamento de Energía de EE. UU. han desarrollado un nuevo marco de aprendizaje automático (ML) que puede concentrarse en qué pasos de una conversión química de varios pasos deben modificarse para mejorar la productividad. El enfoque podría ayudar a guiar el diseño de catalizadores:"negociadores" químicos que aceleran las reacciones.
El equipo desarrolló el método para analizar la conversión de monóxido de carbono (CO) en metanol utilizando un catalizador a base de cobre. La reacción consta de siete pasos elementales bastante sencillos.
"Nuestro objetivo era identificar qué paso elemental en la red de reacción o qué subconjunto de pasos controla la actividad catalítica", dijo Wenjie Liao, el primer autor de un artículo que describe el método que se acaba de publicar en la revista Catalysis Science &Technology . Liao es un estudiante graduado de la Universidad de Stony Brook que ha estado trabajando con científicos en el grupo de estructura y reactividad de catálisis (CRS) en la división de química de Brookhaven Lab.
Ping Liu, el químico de CRS que dirigió el trabajo, dijo:"Usamos esta reacción como un ejemplo de nuestro método de marco ML, pero puede poner cualquier reacción en este marco en general".
Apuntar a las energías de activación
Imagina una reacción química de varios pasos como una montaña rusa con colinas de diferentes alturas. La altura de cada colina representa la energía necesaria para pasar de un paso al siguiente. Los catalizadores reducen estas "barreras de activación" facilitando la unión de los reactivos o permitiéndoles hacerlo a temperaturas o presiones más bajas. Para acelerar la reacción general, un catalizador debe enfocarse en el paso o los pasos que tienen el mayor impacto.
Tradicionalmente, los científicos que buscaban mejorar tal reacción calcularían cómo cambiar cada barrera de activación una a la vez podría afectar la tasa de producción general. Este tipo de análisis podría identificar qué paso fue "limitante de la velocidad" y qué pasos determinan la selectividad de la reacción, es decir, si los reactivos proceden al producto deseado o por un camino alternativo a un subproducto no deseado.

El químico de Brookhaven Lab, Ping Liu, y Wenjie Liao, estudiante de posgrado de la Universidad de Stony Brook, desarrollaron un marco de aprendizaje automático para identificar qué pasos de reacción química podrían ser el objetivo para mejorar la productividad de la reacción. Crédito:Laboratorio Nacional de Brookhaven
Pero, según Liu, "Estas estimaciones terminan siendo muy aproximadas con muchos errores para algunos grupos de catalizadores. Eso realmente ha perjudicado el diseño y la selección de catalizadores, que es lo que estamos tratando de hacer", dijo.
El nuevo marco de aprendizaje automático está diseñado para mejorar estas estimaciones para que los científicos puedan predecir mejor cómo los catalizadores afectarán los mecanismos de reacción y la producción química.
"Ahora, en lugar de mover una barrera a la vez, estamos moviendo todas las barreras simultáneamente. Y usamos el aprendizaje automático para interpretar ese conjunto de datos", dijo Liao.
Este enfoque, dijo el equipo, brinda resultados mucho más confiables, incluso sobre cómo los pasos en una reacción funcionan juntos.
"Bajo las condiciones de reacción, estos pasos no están aislados ni separados entre sí; todos están conectados", dijo Liu. "Si solo haces un paso a la vez, pierdes mucha información:las interacciones entre los pasos elementales. Eso es lo que se ha capturado en este desarrollo", dijo.
Construyendo el modelo
Los científicos comenzaron construyendo un conjunto de datos para entrenar su modelo de aprendizaje automático. El conjunto de datos se basó en los cálculos de la "teoría funcional de la densidad" (DFT) de la energía de activación requerida para transformar una disposición de átomos en la siguiente a través de los siete pasos de la reacción. Luego, los científicos realizaron simulaciones basadas en computadora para explorar qué sucedería si cambiaran las siete barreras de activación simultáneamente:algunas subiendo, otras bajando, algunas individualmente y otras en pares.
"El rango de datos que incluimos se basó en la experiencia previa con estas reacciones y este sistema catalítico, dentro del interesante rango de variación que probablemente le brinde un mejor rendimiento", dijo Liu.
Al simular variaciones en 28 "descriptores", incluidas las energías de activación para los siete pasos más pares de pasos que cambian de dos en dos, el equipo produjo un conjunto de datos completo de 500 puntos de datos. Este conjunto de datos predijo cómo todos esos ajustes individuales y pares de ajustes afectarían la producción de metanol. Luego, el modelo calificó los 28 descriptores según su importancia para impulsar la producción de metanol.
"Nuestro modelo 'aprendió' de los datos e identificó seis descriptores clave que predice que tendrían el mayor impacto en la producción", dijo Liao.
Después de que se identificaron los descriptores importantes, los científicos volvieron a entrenar el modelo ML usando solo esos seis descriptores "activos". Este modelo ML mejorado pudo predecir la actividad catalítica basándose únicamente en los cálculos de DFT para esos seis parámetros.
"En lugar de tener que calcular los 28 descriptores completos, ahora puede calcular solo con los seis descriptores y obtener las tasas de conversión de metanol que le interesan", dijo Liu.
El equipo dice que también pueden usar el modelo para detectar catalizadores. Si pueden diseñar un catalizador que mejore el valor de los seis descriptores activos, el modelo predice una tasa máxima de producción de metanol.
Comprender los mecanismos
Cuando el equipo comparó las predicciones de su modelo con el rendimiento experimental de su catalizador y el rendimiento de aleaciones de varios metales con cobre, las predicciones coincidieron con los hallazgos experimentales. Las comparaciones del enfoque ML con el método anterior utilizado para predecir el rendimiento de las aleaciones demostraron que el método ML es muy superior.
Los datos también revelaron muchos detalles sobre cómo los cambios en las barreras de energía podrían afectar el mecanismo de reacción. De particular interés e importancia fue cómo los diferentes pasos de la reacción funcionan juntos. Por ejemplo, los datos mostraron que, en algunos casos, reducir la barrera energética en el paso de limitación de la tasa por sí solo no mejoraría la producción de metanol. Pero ajustar la barrera de energía de un paso anterior en la red de reacción, mientras se mantiene la energía de activación del paso que limita la velocidad dentro de un rango ideal, aumentaría la producción de metanol.
"Nuestro método nos brinda información detallada que podríamos usar para diseñar un catalizador que coordine bien la interacción entre estos dos pasos", dijo Liu.
Pero Liu está más entusiasmado con el potencial de aplicar dichos marcos de ML basados en datos a reacciones más complicadas.
"Usamos la reacción del metanol para demostrar nuestro método. Pero la forma en que genera la base de datos y cómo entrenamos el modelo ML y cómo interpolamos el papel de la función de cada descriptor para determinar el peso general en términos de su importancia, eso puede ser aplicado fácilmente a otras reacciones", dijo. Descubrimiento de un nuevo catalizador para la hidrogenación selectiva y altamente activa de dióxido de carbono a metanol