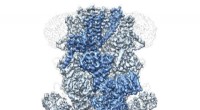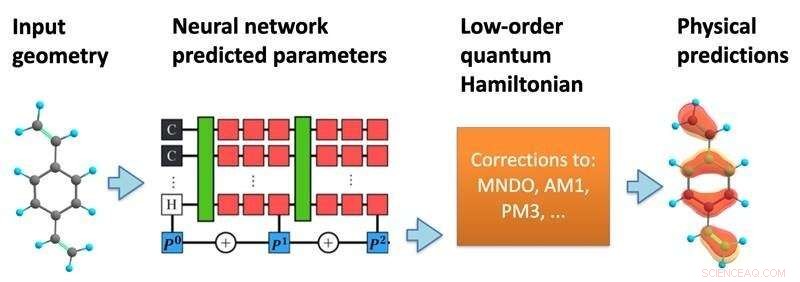
La estructura del modelo. Una red neuronal procesa una geometría molecular para predecir un hamiltoniano cuántico semiempírico, que luego se resuelve de manera autoconsistente para predecir una variedad de propiedades químicas. Crédito:Kipton Barros, Laboratorio Nacional de Los Álamos.
En un nuevo estudio, publicado en Proceedings of the National Academy of Sciences , investigadores del Laboratorio Nacional de Los Alamos han propuesto incorporar más matemáticas de la mecánica cuántica en la estructura de las predicciones de aprendizaje automático. Usando las posiciones específicas de los átomos dentro de una molécula, el modelo de aprendizaje automático predice una matriz hamiltoniana efectiva, que describe los diversos estados electrónicos posibles junto con sus energías asociadas.
En comparación con las simulaciones de química cuántica tradicionales, el enfoque basado en el aprendizaje automático hace predicciones a un costo computacional muy reducido. Permite predicciones cuantitativamente precisas con respecto a las propiedades de los materiales, permite una comprensión interpretable de la naturaleza de los enlaces químicos entre los átomos y puede usarse para predecir otros fenómenos complejos, como la forma en que el sistema responderá a las perturbaciones, como las interacciones luz-materia. El método también proporciona una precisión muy mejorada en relación con los modelos tradicionales de aprendizaje automático y demuestra éxito en la transferibilidad, es decir, la capacidad del modelo para hacer predicciones que van mucho más allá de los datos que formaron la base de su entrenamiento.
Las ecuaciones de la mecánica cuántica proporcionan una hoja de ruta para predecir las propiedades de los productos químicos a partir de teorías científicas básicas. Sin embargo, estas ecuaciones pueden volverse demasiado costosas rápidamente en términos de tiempo y energía de la computadora cuando se usan para predecir el comportamiento en sistemas grandes. El aprendizaje automático ofrece un enfoque prometedor para acelerar este tipo de simulaciones a gran escala. El uso del aprendizaje automático para predecir las propiedades químicas tiene el potencial de grandes avances tecnológicos, con aplicaciones que van desde energía más limpia hasta un diseño de fármacos más rápido. Esta es un área de investigación muy activa, pero la mayoría de los enfoques existentes utilizan enfoques simples y heurísticos para el diseño de los modelos de aprendizaje automático.
En su estudio, los investigadores han demostrado que los modelos de aprendizaje automático pueden imitar la estructura básica de las leyes fundamentales de la naturaleza. Estas leyes pueden ser muy difíciles de simular directamente. El enfoque de aprendizaje automático permite realizar predicciones fáciles de calcular y precisas en una amplia variedad de sistemas químicos.
El modelo de aprendizaje automático mejorado puede predecir de forma rápida y precisa una amplia gama de propiedades de las moléculas. Estos enfoques obtienen muy buenos puntajes en puntos de referencia importantes en química computacional y muestran cómo los métodos de aprendizaje profundo pueden continuar mejorando al incorporar más datos de experimentos. El modelo también puede tener éxito en tareas desafiantes, como predecir la dinámica del estado excitado:cómo se comportan los sistemas con niveles elevados de energía. Esta herramienta es una capacidad revolucionaria para la química cuántica. Permitirá a los investigadores comprender mejor la reactividad y los estados excitados de nuevas moléculas. Las computadoras sobresalen en la clase de química