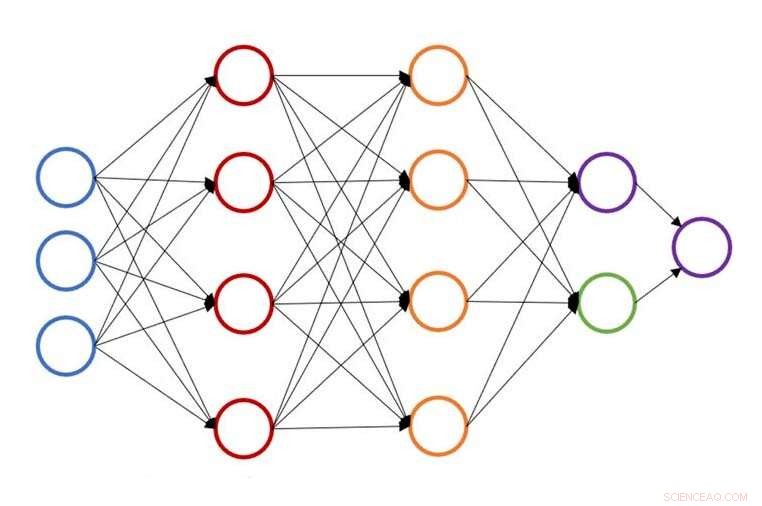
Ilustración de una estructura de red neuronal única para la predicción de energía atómica. Crédito:John Kitchin, Universidad de Carnegie mellon
Aprendizaje automático, un método de análisis de datos utilizado para automatizar la construcción de modelos analíticos, ha reformado la forma en que los científicos e ingenieros llevan a cabo sus investigaciones. Una rama de la inteligencia artificial (IA) y la informática, el método se basa en una gran cantidad de algoritmos y amplios conjuntos de datos para identificar patrones y tomar decisiones de investigación importantes.
Las aplicaciones de las técnicas de aprendizaje automático están surgiendo en el área de la catálisis superficial, permitiendo simulaciones más extensas de nanopartículas, estudios de segregación, optimización de la estructura, aprendizaje sobre la marcha de campos de fuerza, y cribado de alto rendimiento. Sin embargo, trabajar con grandes cantidades de datos a menudo puede ser una tarea larga y costosa desde el punto de vista computacional.
Optimización de geometría, a menudo el paso limitante de la velocidad en las simulaciones moleculares, es una parte clave de los materiales computacionales y la ciencia de superficies. Permite a los investigadores encontrar estructuras atómicas en estado fundamental y vías de reacción, propiedades utilizadas para estimar las propiedades cinéticas y termodinámicas de estructuras moleculares y cristalinas. Si bien es vital, el proceso puede ser relativamente lento, requiriendo una gran cantidad de cálculos para completar.
En la Universidad Carnegie Mellon, John Kitchin está trabajando para acelerar ese proceso proporcionando un método de aprendizaje activo basado en redes neuronales que acelera la optimización geométrica para múltiples configuraciones simultáneamente. El nuevo modelo reduce el número de cálculos de la teoría funcional de la densidad (DFT) o la teoría del medio efectivo (EMT) entre un 50 y un 90 por ciento, permitiendo a los investigadores hacer el mismo trabajo en menos tiempo o más trabajo en la misma cantidad de tiempo.
"Normalmente, cuando hacemos optimización de geometría, empezamos de cero, "dijo Kitchin." Los cálculos rara vez se benefician de algo que sabíamos en el pasado ".
"Al agregar un modelo sustituto al proceso, lo hemos habilitado para que se base en cálculos anteriores, en lugar de empezar desde cero cada vez ".
El estudio ilustra la aceleración en varios estudios de caso, incluidas las superficies con adsorbatos, superficies de metal desnudo, y banda elástica empujada para dos reacciones. En cada caso, el paquete Python del optimizador del entorno de simulación atómica (ASE) permitía realizar menos cálculos de DFT que el método estándar.
El paquete Python ASE-optimizer se ha puesto a disposición de otros ingenieros y científicos para facilitar el uso del aprendizaje activo de conjuntos de redes neuronales para la optimización de la geometría.