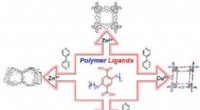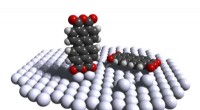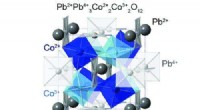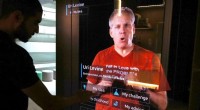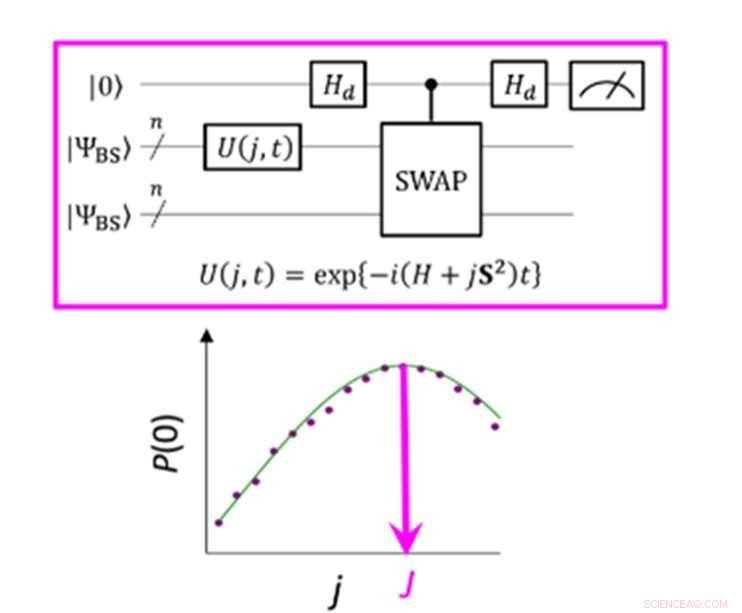
Un circuito cuántico que permite la máxima probabilidad de P (0) en la medición del parámetro J. Crédito:K. Sugisaki, K. Sato y T. Takui
Investigadores de la Universidad de la Ciudad de Osaka utilizan estados de superposición cuántica e inferencia bayesiana para crear un algoritmo cuántico, fácilmente ejecutable en computadoras cuánticas, que calcula de forma precisa y directa las diferencias de energía entre el suelo electrónico y los estados de espín excitado de los sistemas moleculares en tiempo polinomial.
Comprender cómo funciona el mundo natural nos permite imitarlo en beneficio de la humanidad. Piense en cuánto dependemos de las baterías. Lo fundamental es comprender las estructuras moleculares y el comportamiento de los electrones dentro de ellas. El cálculo de las diferencias de energía entre el suelo electrónico de una molécula y los estados de espín excitado nos ayuda a comprender cómo utilizar mejor esa molécula en una variedad de sustancias químicas, aplicaciones biomédicas e industriales. Hemos avanzado mucho en moléculas con sistemas de capa cerrada, en el que los electrones están apareados y son estables. Sistemas de caparazón abierto, por otra parte, son menos estables y su comportamiento electrónico subyacente es complejo, y por tanto más difícil de entender. Tienen electrones desapareados en su estado fundamental, que hacen que su energía varíe debido a la naturaleza intrínseca de los espines de los electrones, y dificulta las mediciones, especialmente a medida que las moléculas aumentan de tamaño y complejidad. Aunque estas moléculas son abundantes en la naturaleza, hay una falta de algoritmos que puedan manejar esta complejidad. Uno de los obstáculos ha sido lidiar con lo que se llama la explosión exponencial del tiempo computacional. Usar una computadora convencional para calcular cómo los espines no apareados influyen en la energía de una molécula de capa abierta llevaría cientos de millones de años, tiempo que los humanos no tienen.
Las computadoras cuánticas están en desarrollo para ayudar a reducir esto a lo que se llama "tiempo polinomial". Sin embargo, el proceso que los científicos han estado usando para calcular las diferencias de energía de las moléculas de capa abierta ha sido esencialmente el mismo para las computadoras convencionales y cuánticas. Esto dificulta el uso práctico de la computación cuántica en aplicaciones químicas e industriales.
"Los enfoques que invocan verdaderos algoritmos cuánticos nos ayudan a tratar los sistemas de capa abierta de manera mucho más eficiente que utilizando computadoras clásicas, "afirman Kenji Sugisaki y Takeji Takui de la Universidad de la ciudad de Osaka. Con sus colegas, desarrollaron un algoritmo cuántico ejecutable en computadoras cuánticas, que puede, por primera vez, calcular con precisión las diferencias de energía entre el suelo electrónico y los estados de espín excitado de los sistemas moleculares de capa abierta. Sus hallazgos fueron publicados en la revista Ciencia química el 24 de diciembre de 2020.
La diferencia de energía entre los estados de espín molecular se caracteriza por el valor del parámetro de interacción de intercambio J. Los algoritmos cuánticos convencionales han podido calcular con precisión las energías para moléculas de capa cerrada "pero no han podido manejar sistemas con una fuerte configuración múltiple personaje, "afirma el grupo. Hasta ahora, Los científicos han asumido que para obtener el parámetro J, primero se debe calcular la energía total de cada estado de giro. En las moléculas de capa abierta esto es difícil porque la energía total de cada estado de giro varía mucho a medida que la molécula cambia en actividad y tamaño. Sin embargo, "la diferencia de energía en sí misma no depende en gran medida del tamaño del sistema, ", señala el equipo de investigación. Esto los llevó a crear un algoritmo con cálculos que se centraban en la diferencia de espín, no los estados de giro individuales. La creación de un algoritmo de este tipo requería que abandonaran las suposiciones desarrolladas a partir de años de uso de computadoras convencionales y se centraran en las características únicas de la computación cuántica, a saber, los "estados de superposición cuántica".
La "superposición" permite que los algoritmos representen dos variables a la vez, lo que permite a los científicos centrarse en la relación entre estas variables sin necesidad de determinar primero sus estados individuales. El equipo de investigación utilizó algo llamado función de onda de simetría rota como una superposición de funciones de onda con diferentes estados de espín y la reescribió en la ecuación hamiltoniana para el parámetro J. Al ejecutar este nuevo circuito cuántico, el equipo pudo concentrarse en las desviaciones de su objetivo y al aplicar la inferencia bayesiana, una técnica de aprendizaje automático, trajeron estas desviaciones para determinar el parámetro de interacción de intercambio J. "Se realizaron simulaciones numéricas basadas en este método para la disociación covalente de hidrógeno molecular (H 2 ), la disociación de triple enlace del nitrógeno molecular (N 2 ), y los estados fundamentales de C, Oh Átomos de Si y NH, OH + , CH 2 , NF y O 2 moléculas con un error de menos de 1 kcal / mol, ", añade el equipo de investigación.
"Planeamos instalar nuestra calculadora de parámetros de acoplamiento Bayesian eXchange con software de funciones de onda de simetría rota (BxB) en computadoras cuánticas a corto plazo equipadas con dispositivos cuánticos (dispositivos NISQ) de escala intermedia (varios cientos de qubits) ruidosos (sin corrección de errores cuánticos) ), probando la utilidad de los cálculos químicos cuánticos de sistemas moleculares de tamaño real ".