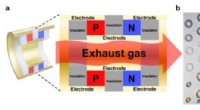
Modelo atómico para la unión de la proteína SARS-CoV-2 S al receptor ACE2 en la membrana de la célula huésped. Crédito:Universidad de California, Berkeleky; Universidad Técnica de Estambul
El virus que está causando estragos en nuestras vidas es una eficiente máquina de infección. Compuesto por solo 29 proteínas (en comparación con nuestras 400, 000), con un genoma 1/200, 000 del tamaño del nuestro, El SARS-CoV-2 ha sido desarrollado de manera experta para engañar a nuestras células para que contribuyan con su maquinaria para ayudar en su propagación.
En los últimos meses, Los científicos han aprendido mucho sobre la mecánica de este enemigo sin sentido. Pero lo que hemos aprendido todavía palidece en comparación con lo que no sabemos.
Hay varias formas en que los científicos descubren el funcionamiento de un virus. Solo utilizando estos métodos en conjunto podemos encontrar y explotar los puntos débiles del coronavirus, dice Ahmet Yildiz, profesor asociado de Física y Biología Celular Molecular en la Universidad de California, Berkeley.
Yildiz y su colaborador Mert Gur en la Universidad Técnica de Estambul están combinando simulaciones de dinámica molecular impulsadas por supercomputadoras con experimentos de una sola molécula para descubrir los secretos del virus. En particular, están estudiando su proteína pico (S), la parte del virus que se une a las células humanas y comienza el proceso de insertar ARN viral en la célula.
"Muchos grupos están atacando diferentes etapas de este proceso, ", Dijo Gur." Nuestro objetivo inicial es utilizar simulaciones de dinámica molecular para identificar los procesos que suceden cuando el virus se une a la célula huésped ".
Hay tres fases críticas que permiten que la proteína espiga ingrese a la célula y comience a replicarse. Dice Yildiz.
Primero, la proteína de pico necesita transformarse de una configuración cerrada a una abierta. Segundo, la proteína de pico se une a su receptor en el exterior de nuestras células. Esta unión desencadena un cambio conformacional dentro de la proteína de la espiga y permite que otra proteína humana rompa la espiga. Finalmente, la superficie recién expuesta de la espiga interactúa con la membrana de la célula huésped y permite que el ARN viral ingrese y se apropie de la célula.
A principios de febrero Las imágenes de microscopio electrónico revelaron la estructura de la proteína de pico. Pero las instantáneas solo mostraron las configuraciones principales que toma la proteína, no el transicional, pasos intermedios. "Solo vemos instantáneas de conformaciones estables, "Yildiz dijo." Debido a que no conocemos el momento de los eventos que permiten que la proteína pase de una conformación estable a la siguiente, todavía no conocemos esas conformaciones intermedias ".
Ahí es donde entra en juego el modelado por computadora. Las imágenes microscópicas proporcionan un punto de partida útil para crear modelos de cada átomo de la proteína, y su entorno (agua, iones, y los receptores de la célula). Desde allí, Yildiz y Gur pusieron la proteína en movimiento y observaron para ver qué sucedía.
"Demostramos que la proteína S visita un estado intermedio antes de que pueda acoplarse a la proteína receptora en la membrana de la célula huésped", dijo Gur. "Este estado intermedio puede ser útil para la selección de fármacos para evitar que la proteína S inicie una infección viral".
Mientras que muchos otros grupos en todo el mundo están investigando el bolsillo de unión del virus, con la esperanza de encontrar un fármaco que pueda impedir que el virus se adhiera a las células humanas, Yildiz y Gur están adoptando un enfoque más matizado.
"La proteína de pico se une fuertemente a su receptor con una compleja red de interacción, ", Explicó Yildiz." Demostramos que si rompes una de esas interacciones, todavía no podrá detener el enlace. Es por eso que algunos de los estudios básicos de desarrollo de fármacos pueden no producir los resultados deseados ".
Pero si es posible evitar que la proteína de pico pase de un estado cerrado a un estado abierto, o un tercero, estado intermedio del que ni siquiera somos conscientes del estado abierto, que podría prestarse a un tratamiento.
Encontrar, y romper los lazos importantes
El segundo uso de simulaciones por computadora por parte de Yildiz y Gur identificó no solo nuevos estados, sino los aminoácidos específicos que estabilizan cada estado.
"Si podemos determinar los vínculos importantes a nivel de un solo aminoácido, que interacciones se estabilizan y son críticas para estas confirmaciones, es posible que apunte a esos estados con moléculas pequeñas, "Dijo Yildiz.
Simular este comportamiento a nivel del átomo o del aminoácido individual es increíblemente intensivo desde el punto de vista informático. A Yildiz y Gur se les concedió tiempo en la supercomputadora Stampede2 en el Texas Advanced Computing Center (TACC), la segunda supercomputadora más rápida en una universidad de EE. UU. Y la 19ª más rápida en general, a través del COVID-19 HPC Consortium. Simular un microsegundo del virus y sus interacciones con las células humanas, aproximadamente un millón de átomos en total, lleva semanas en una supercomputadora ... y llevaría años sin una.
"Es un proceso computacionalmente exigente, ", Dijo Yildiz." Pero el poder predictivo de este enfoque es muy poderoso ".
Equipo de Yildiz y Gur, junto con aproximadamente otros 40 grupos de investigación que estudian COVID-19, se les ha dado acceso prioritario a los sistemas TACC. "No estamos limitados por la velocidad a la que ocurren las simulaciones, por lo que existe una carrera en tiempo real entre nuestra capacidad para ejecutar simulaciones y analizar los datos ".
Con el tiempo de la esencia Gur y sus colaboradores han realizado cálculos, recreando las peregrinaciones atómicas de la proteína pico a medida que se acerca, se une a, e interactúa con los receptores de la enzima convertidora de angiotensina 2 (ACE2), proteínas que recubren la superficie de muchos tipos de células.
Sus hallazgos iniciales, que propuso la existencia de un estado intermedio semiabierto de la proteína S compatible con la unión de RBD-ACE2 a través de simulaciones de dinámica molecular (MD) de todos los átomos, fue publicado en el Revista de física química .
Es más, mediante la realización de simulaciones MD de todos los átomos, identificaron una extensa red de puentes de sal, interacciones hidrofóbicas y electrostáticas, y enlaces de hidrógeno entre el dominio de unión al receptor de la proteína de pico y ACE2. Los resultados de estos hallazgos se publicaron en BioRxiv.
La mutación de los residuos en el dominio de unión al receptor no fue suficiente para desestabilizar la unión, pero redujo el trabajo promedio para desvincular la proteína de pico de ACE2. Proponen que el bloqueo de este sitio a través de un anticuerpo neutralizante o nanocuerpo podría resultar una estrategia eficaz para inhibir las interacciones de la proteína de pico-ACE2.
Para confirmar que la información obtenida por computadora es precisa, El equipo de Yildiz realizó experimentos de laboratorio utilizando transferencia de energía de resonancia de fluorescencia de una sola molécula (o smFRET), una técnica biofísica utilizada para medir distancias en la escala de uno a 10 nanómetros en moléculas individuales
"La técnica nos permite ver los cambios conformacionales de la proteína midiendo la transferencia de energía entre dos sondas emisoras de luz, "Dijo Yildiz.
Aunque los científicos todavía no tienen una técnica para ver los detalles atómicos de las moléculas en movimiento en tiempo real, la combinación de microscopía electrónica, formación de imágenes de una sola molécula, y las simulaciones por computadora pueden proporcionar a los investigadores una imagen rica del comportamiento del virus, Dice Yildiz.
"Podemos obtener instantáneas de resolución atómica de moléculas congeladas usando microscopía electrónica. Podemos obtener simulaciones a nivel atómico de la proteína en movimiento usando dinámica molecular en una escala de tiempo corta. Y usando técnicas de una sola molécula podemos derivar la dinámica que falta en el electrón microscopía y simulaciones, Yildiz concluyó:"La combinación de estos métodos nos da una imagen completa y disecciona el mecanismo de entrada de un virus en la célula huésped".