Crédito:Elena Khavina / MIPT
Investigadores del Instituto de Física y Tecnología de Moscú han publicado una revisión sobre cristalografía de femtosegundos en serie, uno de los métodos más prometedores para analizar la estructura terciaria de las proteínas. Esta técnica ha evolucionado rápidamente durante la última década, abriendo nuevas perspectivas para el diseño racional de fármacos dirigidos a proteínas previamente inaccesibles al análisis estructural. El artículo salió en la revista Opinión de expertos sobre el descubrimiento de fármacos .
Cristalografía de rayos X
La cristalografía de rayos X es uno de los principales métodos para revelar la estructura tridimensional de las macromoléculas biológicas, como las proteínas. Ha ayudado a determinar la estructura de numerosas enzimas y receptores farmacológicamente importantes, permitiendo el diseño de fármacos dirigidos a estas proteínas.
El método consiste en cristalizar una proteína y estudiarla mediante difracción de rayos X. Primero, la proteína se aísla y se purifica. Luego, el solvente se seca gradualmente. Como resultado, las moléculas cuya estructura se investiga forman cristales, caracterizado por un orden interno. Al exponer un cristal a rayos X en un dispositivo especial, los investigadores obtienen un patrón de difracción. Contiene información sobre las posiciones de los átomos en el cristal. Un análisis detallado del patrón revela la estructura tridimensional de las moléculas de proteína constituyentes.
Antes de la llegada de este método, Los nuevos fármacos se buscaron principalmente de forma empírica:ya sea cambiando la estructura de las moléculas que se sabe que afectan a la proteína diana, o clasificando matrices de moléculas en bibliotecas químicas. Ahora que las estructuras tridimensionales de muchas proteínas diana están disponibles, los investigadores pueden verlos en la pantalla de una computadora y clasificar rápidamente millones de compuestos en busca de fármacos candidatos. De esa manera, ahorran mucho tiempo y dinero gastado anteriormente en síntesis química y experimentos "húmedos".
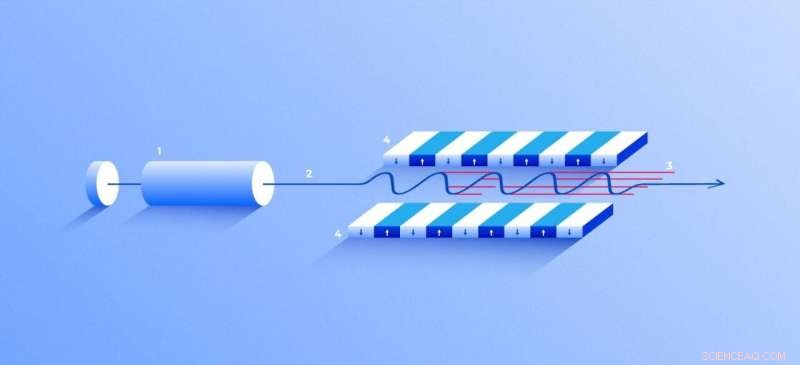
Láser de rayos X de electrones libres. Una fuente (1) emite electrones libres (2) moviéndose simplemente decenas de veces más lento que la velocidad de la luz a través del ondulador (4), un túnel revestido de muchos imanes. El campo magnético hace que un electrón que viaja a través del túnel oscile y, por lo tanto, emita rayos X. El movimiento de los electrones en el ondulador se sincroniza para generar tensiones, pulsos de rayos X de alta frecuencia de notable intensidad (3). Crédito:Elena Khavina / MIPT
La cristalografía de rayos X produce buenos resultados para cristales grandes, estable, y homogéneo, es decir, sin impurezas ni defectos estructurales. Para detectar mejor una señal de difracción débil, se necesita un potente pulso de radiación, pero no tan poderoso como para destruir el cristal. En cristalografía de rayos X convencional, un cristal de proteína se hace girar en el haz de rayos X para producir patrones de difracción para diversas orientaciones espaciales. Esto captura la máxima información sobre la estructura.
Método para objetivos difíciles
Poco después de que surgiera la cristalografía de rayos X, quedó claro que no todas las macromoléculas biológicas pueden cristalizarse. Algunas proteínas normalmente se disuelven en el medio celular interno. Por lo que es bastante fácil ponerlos en solución, evaporarlo, y obtener un gran cristal regular. Pero las proteínas de membrana, muchos receptores entre ellos, forman cristales que no son lo suficientemente grandes y puros para la cristalografía de rayos X estándar. Dicho eso muchas de estas proteínas están involucradas en el desarrollo de enfermedades, lo que significa que su estructura es de gran interés para los farmacólogos.
Hace menos de una década, se encontró una solución para las proteínas de membrana. Esta nueva técnica, llamada cristalografía de rayos X en serie de femtosegundos, o SFX, se basa en láseres de rayos X de electrones libres, desarrollado poco antes de SFX.
Alexey Mishin, subdirector del Laboratorio de Biología Estructural de Receptores del MIPT, quién fue el coautor del estudio, explicó:"Lo que la convierte en una tecnología innovadora es una densidad de energía muy alta del pulso láser. El objeto está expuesto a una radiación tan poderosa que se desmorona, inevitable y casi instantáneamente. Pero antes de que lo haga algunos cuantos individuales del pulso láser se dispersan fuera de la muestra y terminan en el detector. Este es el llamado principio de difracción antes de la destrucción para estudiar la estructura de la proteína original ".

Dos formas de introducir cristales en el área de operación del dispositivo:en una corriente de líquido (3, izquierda) y sobre un sustrato sólido (3, Derecha). En ambos casos, el haz de rayos X (2) que atraviesa los cristales genera un patrón de difracción en la pantalla (1). Crédito:Elena Khavina / MIPT
Los láseres de rayos X de electrones libres resultaron útiles fuera de la biología:en los últimos años, SFX ha sido utilizado cada vez más por físicos y químicos, también. El primer dispositivo estuvo disponible para los experimentadores en 2009, y ahora hay cinco centros abiertos a investigadores en los EE. UU., Japón, Corea del Sur, Alemania, y Suiza. Se está construyendo uno nuevo en China, y la instalación de EE. UU., históricamente la primera, ha anunciado planes de modernización.
Si bien la nueva tecnología ha ofrecido a los investigadores un vistazo a la estructura de las proteínas que anteriormente eludían el análisis, también ha promovido soluciones técnicas y matemáticas novedosas. La cristalografía de rayos X convencional implica exponer un cristal a la radiación desde varios ángulos y analizar colectivamente los patrones de difracción resultantes. En SFX, el cristal se destruye instantáneamente por la primera interacción con un poderoso pulso de rayos X. Por lo tanto, los investigadores deben repetir el proceso con muchos cristales pequeños y analizar los datos "en serie" así generados. de ahí el nombre del método.
Otro desafío es seleccionar las muestras para SFX. En cristalografía de rayos X convencional, simplemente elegir el cristal más grande y de mayor calidad era el camino a seguir. Esto se puede hacer manualmente, observando las muestras disponibles. El nuevo procedimiento requiere trabajar con una suspensión de muchos cristales pequeños de diferente tamaño y calidad. Se utilizan centrífugas y filtros con dimensiones de poro conocidas para separar los cristales por tamaño.
Debían elaborarse métodos para colocar las muestras en la cámara, también. Los láseres de rayos X de electrones libres tienen una cierta frecuencia máxima a la que pueden emitir pulsos de radiación. Para reducir los gastos y el consumo de tiempo, Los cristales nuevos deben introducirse en la cámara con la misma frecuencia. Hasta aquí, Se han desarrollado dos enfoques para hacer esto. Debajo del primero, los cristales entran en la cámara en suspensión líquida, suministrado por un inyector. El chorro que sale del inyector es "exprimido" por una corriente de gas para asegurar la correcta colocación de la muestra. Es decir, al pasar, un cristal termina precisamente en el centro del rayo láser (fig.2, izquierda). Alternativamente, los cristales de proteína pueden extenderse sobre un sustrato transparente a los rayos X y alimentarse automáticamente al rayo láser antes de cada pulso (fig.2, Derecha).
Desde que produjo sus primeros resultados en 2011, SFX ha revelado más de 200 estructuras de proteínas. Entre ellos se encuentran 51 objetivos potencialmente importantes para la farmacología:receptores de membrana, fermentos, proteínas virales, etc., que antes eran inaccesibles a las técnicas analíticas convencionales.
La revisión sistemática de la tecnología aplicada a la biología y la farmacología por parte del equipo de MIPT sin duda ayudará a otros investigadores que buscan obtener las estructuras de los fármacos objetivos clave para desarrollar nuevos medicamentos.