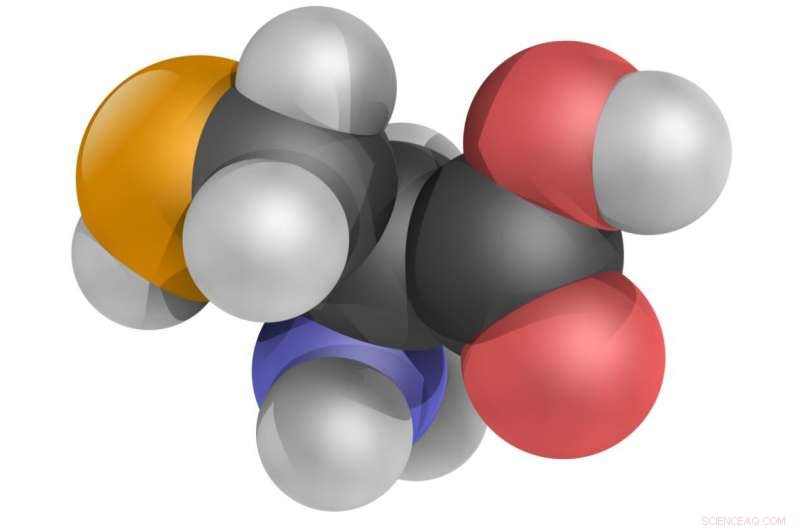
El aminoácido selenocisteína, Modelo de bolas 3D. Crédito:YassineMrabet / CC BY 3.0 / Wikipedia
Casi todos los procesos biológicos fundamentales necesarios para la vida son llevados a cabo por proteínas. Crean y mantienen las formas de células y tejidos; constituyen las enzimas que catalizan las reacciones químicas que sustentan la vida; actuar como fábricas moleculares, transportadores y motores; servir como señal y receptor para comunicaciones celulares; y mucho más.
Compuesto por largas cadenas de aminoácidos, las proteínas realizan estas innumerables tareas al plegarse en estructuras tridimensionales precisas que gobiernan cómo interactúan con otras moléculas. Debido a que la forma de una proteína determina su función y la extensión de su disfunción en la enfermedad, Los esfuerzos para iluminar las estructuras de las proteínas son fundamentales para toda la biología molecular y, en particular, ciencia terapéutica y desarrollo de medicamentos que salvan y alteran la vida.
En años recientes, Los métodos computacionales han logrado avances significativos en la predicción de cómo se pliegan las proteínas basándose en el conocimiento de su secuencia de aminoácidos. Si se realiza plenamente, estos métodos tienen el potencial de transformar prácticamente todas las facetas de la investigación biomédica. Enfoques actuales, sin embargo, están limitados en la escala y el alcance de las proteínas que se pueden determinar.
Ahora, Un científico de la Escuela de Medicina de Harvard ha utilizado una forma de inteligencia artificial conocida como aprendizaje profundo para predecir la estructura tridimensional de cualquier proteína de manera efectiva basándose en su secuencia de aminoácidos.
Informar en línea en Sistemas celulares el 17 de abril El biólogo de sistemas Mohammed AlQuraishi detalla un nuevo enfoque para determinar computacionalmente la estructura de las proteínas, logrando una precisión comparable a los métodos actuales de vanguardia, pero a velocidades un millón de veces más rápidas.
"El plegamiento de proteínas ha sido uno de los problemas más importantes para los bioquímicos durante el último medio siglo, y este enfoque representa una forma fundamentalmente nueva de abordar ese desafío, "dijo AlQuraishi, instructor en biología de sistemas en el Instituto Blavatnik de HMS y becario en el Laboratorio de Farmacología de Sistemas. "Ahora tenemos una perspectiva completamente nueva desde la que explorar el plegamiento de proteínas, y creo que acabamos de empezar a arañar la superficie ".
Fácil de declarar
Si bien fue muy exitoso, Los procesos que utilizan herramientas físicas para identificar estructuras de proteínas son costosos y requieren mucho tiempo. incluso con técnicas modernas como la microscopía crioelectrónica. Como tal, la gran mayoría de las estructuras proteicas, y los efectos de las mutaciones que causan enfermedades en estas estructuras, aún se desconocen en gran medida.
Los métodos computacionales que calculan cómo se pliegan las proteínas tienen el potencial de reducir drásticamente el costo y el tiempo necesarios para determinar la estructura. Pero el problema es difícil y sigue sin resolverse después de casi cuatro décadas de intenso esfuerzo.
Las proteínas se crean a partir de una biblioteca de 20 aminoácidos diferentes. Estos actúan como letras en un alfabeto, combinando en palabras, oraciones y párrafos para producir un número astronómico de textos posibles. A diferencia de las letras del alfabeto, sin embargo, los aminoácidos son objetos físicos colocados en el espacio tridimensional. A menudo, Las secciones de una proteína estarán en estrecha proximidad física pero estarán separadas por grandes distancias en términos de secuencia, como sus cadenas de aminoácidos forman bucles, espirales, sábanas y giros.
"Lo convincente del problema es que es bastante fácil de enunciar:tome una secuencia y descubra la forma, "Dijo AlQuraishi." Una proteína comienza como una cadena no estructurada que tiene que tomar una forma tridimensional, y el posible conjunto de formas en las que se puede doblar una cuerda es enorme. Muchas proteínas tienen miles de aminoácidos de longitud, y la complejidad excede rápidamente la capacidad de la intuición humana o incluso las computadoras más poderosas ".
Difícil de resolver
Para abordar este desafío, Los científicos aprovechan el hecho de que los aminoácidos interactúan entre sí basándose en las leyes de la física, buscando estados energéticamente favorables como una pelota que rueda cuesta abajo para asentarse en el fondo de un valle.
Los algoritmos más avanzados calculan la estructura de las proteínas ejecutándose en supercomputadoras, o potencia informática de fuentes múltiples en el caso de proyectos como Rosetta @ Home y Folding @ Home, para simular la compleja física de las interacciones de aminoácidos mediante la fuerza bruta. Para reducir los requisitos computacionales masivos, estos proyectos se basan en el mapeo de nuevas secuencias en plantillas predefinidas, que son estructuras proteicas previamente determinadas a través de experimentos.
Otros proyectos, como AlphaFold de Google, han generado un gran entusiasmo reciente al utilizar avances en inteligencia artificial para predecir la estructura de una proteína. Para hacerlo estos enfoques analizan enormes volúmenes de datos genómicos, que contienen el plano de las secuencias de proteínas. Buscan secuencias en muchas especies que probablemente hayan evolucionado juntas, utilizando tales secuencias como indicadores de proximidad física cercana al ensamblaje de la estructura de guía.
Estos enfoques de IA, sin embargo, no prediga estructuras basándose únicamente en la secuencia de aminoácidos de una proteína. Por lo tanto, tienen una eficacia limitada para proteínas para las que no hay conocimiento previo, proteínas evolutivas únicas o proteínas novedosas diseñadas por humanos.
Entrenando profundamente
Para desarrollar un nuevo enfoque, AlQuraishi aplicó el llamado aprendizaje profundo diferenciable de extremo a extremo. Esta rama de la inteligencia artificial ha reducido drásticamente el poder computacional y el tiempo necesario para resolver problemas como el reconocimiento de imágenes y voz. habilitando aplicaciones como Siri de Apple y Google Translate.
En esencia, El aprendizaje diferenciable implica una enorme función matemática, una versión mucho más sofisticada de una ecuación de cálculo de la escuela secundaria, organizada como una red neuronal, con cada componente de la red alimentando información hacia adelante y hacia atrás.
Esta función puede sintonizarse y ajustarse a sí misma, una y otra vez a niveles de complejidad inimaginables, para "aprender" precisamente cómo una secuencia de proteínas se relaciona matemáticamente con su estructura.
AlQuraishi desarrolló un modelo de aprendizaje profundo, denominada red geométrica recurrente, que se centra en las características clave del plegamiento de proteínas. Pero antes de que pueda hacer nuevas predicciones, debe ser entrenado utilizando secuencias y estructuras previamente determinadas.
Para cada aminoácido, el modelo predice el ángulo más probable de los enlaces químicos que conectan el aminoácido con sus vecinos. También predice el ángulo de rotación alrededor de estos enlaces, lo que afecta la forma en que cualquier sección local de una proteína se relaciona geométricamente con la estructura completa.
Esto se hace repetidamente, con cada cálculo informado y refinado por las posiciones relativas de todos los demás aminoácidos. Una vez que se completa toda la estructura, el modelo verifica la precisión de su predicción comparándola con la estructura de la proteína de "verdad fundamental".
Todo este proceso se repite para miles de proteínas conocidas, con el modelo aprendiendo y mejorando su precisión con cada iteración.
Nueva vista
Una vez que su modelo fue entrenado, AlQuraishi probó su poder predictivo. Él comparó su desempeño con otros métodos de varios años recientes de la Evaluación Crítica de la Predicción de la Estructura de la Proteína, un experimento anual que prueba la capacidad de los métodos computacionales para hacer predicciones usando estructuras de proteínas que han sido determinadas pero no publicadas.
Descubrió que el nuevo modelo superó a todos los demás métodos para predecir estructuras de proteínas para las que no existen plantillas preexistentes. incluidos los métodos que utilizan datos coevolutivos. También superó a todos los métodos, excepto a los mejores, cuando las plantillas preexistentes estaban disponibles para hacer predicciones.
Si bien estas ganancias en precisión son relativamente pequeñas, AlQuraishi señala que cualquier mejora en el extremo superior de estas pruebas es difícil de lograr. Y debido a que este método representa un enfoque completamente nuevo para el plegamiento de proteínas, puede complementar los métodos existentes, tanto computacional como físico, para determinar una gama de estructuras mucho más amplia de lo que era posible anteriormente.
Sorprendentemente, el nuevo modelo realiza sus predicciones en alrededor de seis a siete órdenes de magnitud más rápido que los métodos computacionales existentes. Entrenar al modelo puede llevar meses, pero una vez entrenado, puede hacer predicciones en milisegundos en comparación con las horas o los días que toma con otros enfoques. Esta espectacular mejora se debe en parte a la única función matemática en la que se basa, requiriendo solo unos pocos miles de líneas de código de computadora para ejecutarse en lugar de millones.
La rápida velocidad de las predicciones de este modelo permite nuevas aplicaciones que antes eran lentas o difíciles de lograr, AlQuraishi dijo:como predecir cómo las proteínas cambian de forma cuando interactúan con otras moléculas.
"Enfoques de aprendizaje profundo, no solo mio, seguirá creciendo en su poder predictivo y en popularidad, porque representan un mínimo, paradigma simple que puede integrar nuevas ideas más fácilmente que los modelos complejos actuales, "añadió.
El nuevo modelo no está listo para su uso inmediato en, decir, descubrimiento o diseño de fármacos, AlQuraishi dijo:debido a que su precisión actualmente se encuentra en algún lugar alrededor de 6 angstroms, todavía a cierta distancia de los 1 o 2 angstroms necesarios para resolver la estructura atómica completa de una proteína. Pero hay muchas oportunidades para optimizar el enfoque, él dijo, incluyendo más reglas integradoras extraídas de la química y la física.
"La predicción precisa y eficiente del plegamiento de proteínas ha sido un santo grial para el campo, y tengo la esperanza y la expectativa de que este enfoque, combinado con todos los otros métodos notables que se han desarrollado, podrá hacerlo en un futuro próximo, "Dijo AlQuraishi." Podríamos resolver esto pronto, y creo que nadie hubiera dicho eso hace cinco años. Es muy emocionante y también algo impactante al mismo tiempo ".
Para ayudar a otros a participar en el desarrollo de métodos, AlQuraishi ha hecho que su software y sus resultados estén disponibles gratuitamente a través de la plataforma de intercambio de software GitHub.
"Una característica notable del trabajo de AlQuraishi es que un solo investigador, integrado en el rico ecosistema de investigación de la Escuela de Medicina de Harvard y la comunidad biomédica de Boston, puede competir con empresas como Google en una de las áreas más importantes de la informática, "dijo Peter Sorger, HMS Otto Krayer Profesor de Farmacología de Sistemas en el Instituto Blavatnik de HMS, director del Laboratorio de Farmacología de Sistemas en HMS y mentor académico de AlQuraishi.
"No es prudente subestimar el impacto disruptivo de compañeros brillantes como AlQuraishi que trabajan con software de código abierto en el dominio público, "Dijo Sorger.