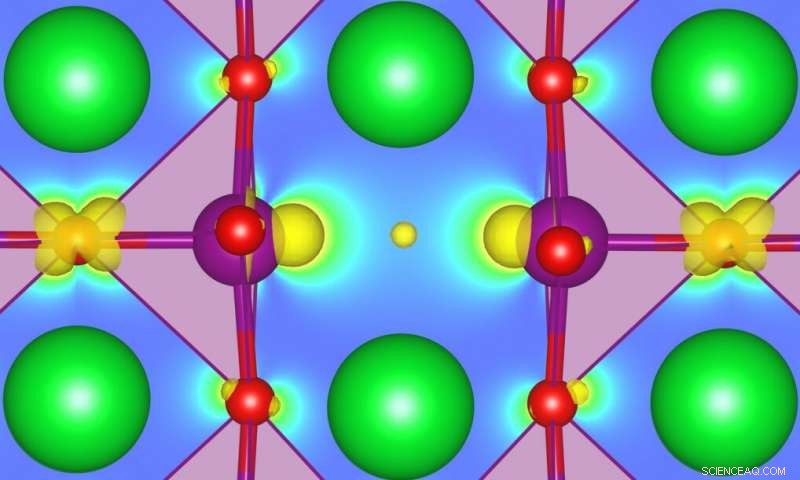
Entender cómo los defectos pueden afectar las propiedades del estado fundamental, promover las transiciones de fase, o habilitar funcionalidades completamente nuevas en algunos óxidos fuertemente correlacionados se ha convertido en un tema de gran interés en el campo del diseño y descubrimiento de nuevos materiales funcionales. SrMnO3 (SMO) es un ejemplo particularmente interesante, pero se necesita una mejor caracterización. Los investigadores de MARVEL han desarrollado ahora un método que puede conducir a predicciones más precisas de la energía de los defectos asociados con los estados dentro del espacio en semiconductores o aislantes. Crédito:Ulrich Aschauer
Entender cómo los defectos pueden afectar las propiedades del estado fundamental, promover las transiciones de fase, o habilitar funcionalidades completamente nuevas en algunos óxidos fuertemente correlacionados se ha convertido en un tema de gran interés en el campo del diseño y descubrimiento de nuevos materiales funcionales. SrMnO 3 (SMO) es un ejemplo particularmente interesante, pero se necesita una mejor caracterización. Los investigadores de MARVEL han desarrollado ahora un método que puede conducir a predicciones más precisas de la energía de los defectos asociados con los estados dentro del espacio en semiconductores o aislantes.
Algunos óxidos de perovskita, por ejemplo, han mostrado un amplio espectro de propiedades funcionales tecnológicamente relevantes, como la ferroelectricidad y el magnetismo, que pueden ajustarse mediante tensión. Cepa, sin embargo, también se acopla con la química del defecto para determinar las propiedades del material.
SrMnO 3 (SMO) es un ejemplo particularmente interesante para examinar la funcionalidad resultante de una interacción compleja de tensión, orden magnético, distorsiones polares, y vacantes de oxígeno que son defectos ubicuos en estos materiales. En particular, La teoría ha predicho que las películas delgadas de SMO pasarán de ser antiferromagnéticas a ferromagnéticas con una creciente deficiencia de oxígeno. que está respaldado por estudios experimentales recientes.
Sin embargo, estas predicciones anteriores se basaron en cálculos de la teoría funcional de la densidad (DFT) que incorporaron una corrección U basada en las propiedades electrónicas y magnéticas de las manganitas estequiométricas. Si bien la inclusión de U, destinada a corregir la auto-interacción de electrones en óxidos complejos, es necesaria en tales materiales, La elección específica de U basada en las propiedades estequiométricas del material podría conducir a posibles deficiencias en la descripción del SMO defectuoso:los iones de manganeso alrededor del defecto tienen un entorno de coordinación diferente.
Dependiendo del estado de carga por defecto, un problema adicional está relacionado con la descripción de múltiples estados de oxidación presentes en SMO defectuoso. La formación de vacantes de oxígeno generalmente se compensa con la carga mediante una reducción del estado de oxidación (OS) de los iones de manganeso adyacentes a la vacante, que, por lo tanto, puede no ser adecuadamente descrito por la misma U.
Esta es la razón por la cual la postdoctorado de la Universidad de Berna, Chiara Ricca y sus colegas, decidieron que era fundamental tener en cuenta los efectos estructurales y químicos locales para cada sitio de metal de transición en el óxido cuando se busca una descripción precisa del SMO defectuoso. En colaboración con un equipo del laboratorio THEOS de Nicola Marzari, que desarrolló recientemente un enfoque basado en la teoría de perturbación funcional de densidad (DFPT) para calcular los parámetros U, utilizaron valores U dependientes del sitio autoconsistentes calculados a partir de los primeros principios para estudiar la química de los defectos y las propiedades magnéticas de las películas delgadas deformadas y a granel de SMO.
"Esta colaboración extremadamente estrecha entre los dos grupos, uno centrado en el desarrollo de métodos y el otro en aplicaciones en materiales de óxido defectuosos, surgió al unir estos diferentes focos de investigación bajo el paraguas de MARVEL ", dijo Ulrich Aschauer de la Universidad de Berna, uno de los dos IP involucrados en el trabajo.
Los resultados muestran que esta U autoconsistente mejora la estructura de SrMnO estequiométrico 3 con respecto a otros métodos, incluyendo uno que usa una U empírica. Para sistemas defectuosos, U cambia en función de la distancia entre el lugar del metal de transición y el defecto, su estado de oxidación, su número de coordinación, y la fase magnética del material. Teniendo en cuenta esta dependencia, Sucesivamente, afecta las energías de formación de defectos calculadas y las transiciones de fase magnética inducidas por deformación y / o defecto predichas, especialmente cuando aparecen estados localizados ocupados en la banda prohibida del material tras la creación del defecto.
"Creemos que este enfoque puede conducir a predicciones más precisas de la energética de los defectos asociados con los estados en el espacio en semiconductores o aisladores, en comparación con la DFT estándar y posiblemente con los funcionales híbridos a un costo computacional que es significativamente más bajo que para el último". "Dijo Ricca." Esto es gracias a una descripción adecuada de los efectos químicos estructurales y locales inducidos por los defectos ".