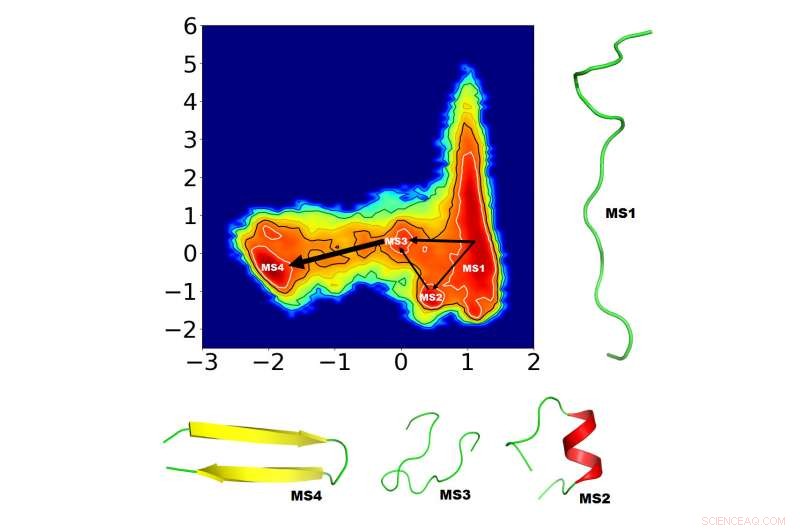
Los científicos buscan comprender mejor el plegamiento de proteínas para curar enfermedades de plegamiento incorrecto, pero este proceso increíblemente complejo requiere algoritmos sofisticados para identificar los mecanismos de plegado. Los biofísicos computacionales han propuesto una nueva forma de identificar los factores más cruciales para el plegamiento de proteínas. Demostraron el breve tiempo de simulación de su enfoque en una proteína pequeña pero intrigante, "Horquilla beta GB1, " en el Revista de física química . Los cuatro nuevos estados de plegado intermedios (MS1-4) identificados por el equipo se muestran aquí, junto con las posibles vías de conexión. El grosor de las flechas de interconexión refleja la probabilidad de que se produzca la vía. Crédito:Navjeet Ahalawat y Jagannath Mondal
Los patrones de plegamiento de una proteína les ayudan a realizar sus tareas específicas. Como verdaderos "hacedores" de la célula, incluso una pequeña alteración en la columna vertebral de los aminoácidos de una proteína puede causar un plegado incorrecto y obstaculizar la funcionalidad de la proteína o causar una enfermedad. Por ejemplo, si tau, una proteína que ayuda a estabilizar la estructura de las células cerebrales, está mal plegado, puede formar tau-enredos, que se ven comúnmente en pacientes con Alzheimer.
Los científicos buscan comprender mejor el plegamiento de proteínas para curar las enfermedades de plegamiento incorrecto, pero este proceso increíblemente complejo requiere algoritmos sofisticados para identificar los mecanismos de plegado. Los biofísicos computacionales del Instituto Tata de Investigación Fundamental de Hyderabad (TIFR-H) han propuesto una nueva forma de identificar los factores más cruciales para el plegamiento de proteínas. Demostraron el breve tiempo de simulación de su enfoque en una proteína pequeña pero intrigante, "GB1 beta-horquilla, " en el Revista de física química , de AIP Publishing.
"Al combinar un método conocido como 'análisis de componentes independientes basado en la estructura del tiempo' (TICA) con simulaciones breves de dinámica molecular, hemos encontrado cuatro estados de plegado intermedios físicamente significativos, no observado previamente, y mostró estados helicoidales que normalmente no pueden ser detectados por otros métodos, "dijo Navjeet Ahalawat, un autor en el papel.
Cada átomo de una proteína se puede plegar en tres dimensiones, pero con millones de átomos presentes incluso en proteínas simples, la tarea de comprender la combinación de plegamiento colectivo se vuelve complicada. Los científicos han considerado los diferentes factores que influyen en el plegamiento de proteínas, como el enlace de hidrógeno, y los combinó en descripciones generales llamadas variables colectivas (CV). Sin embargo, con muchos factores potenciales, los científicos carecen de una buena forma de encontrar CV que describan adecuadamente un proceso factible.
"Hay muchas formas en que las proteínas pueden pasar del estado desplegado al plegado, así que lo más difícil es decidir por dónde empezar, "Ahalawat dijo. Jagannath Mondal, otro autor en el papel, agregó que era fácil "perderse en los datos".
El equipo decidió estudiar la horquilla que sobresale externamente de la proteína GB1 debido a la gran cantidad de trabajo existente y muchas posibilidades potenciales de plegamiento ya estimadas en CV anteriores. Ahalawat y Mondal tomaron varios CV GB1 existentes como CV constituyentes y los combinaron linealmente utilizando TICA para identificar un par de CV "optimizados". Luego, ingresaron los CV optimizados en el modelo de estado de Markov e identificaron cuatro estados de plegamiento intermedios junto con las posibles vías de conexión.
"Preguntamos, ¿Cuáles son las características estimadas anteriormente para esta proteína en particular que realmente podrían desempeñar un papel clave en el sistema? ¿Y podemos encontrar la combinación correcta de condiciones? ", Dijo Ahalawat." En nuestro trabajo ahora podemos decir cuantitativamente si esa característica es relevante para el proceso ".
"Utilizando simulaciones breves, hemos encontrado el peso que realmente necesita usar en una combinación, y esto da el patrón de plegado correcto para una proteína, Mondal añadió. "Es una forma muy barata de averiguar el plegamiento de proteínas".
En su método, Se necesitan datos de estudios previos para identificar CV óptimos. El equipo prevé que su técnica se puede utilizar para descubrir el mecanismo interno del plegamiento de proteínas saludables para corregir enfermedades que causan proteínas mal plegadas. También quieren seguir desarrollando su método de optimización de CV y aplicarlos en el reconocimiento biomolecular y el descubrimiento de fármacos. "En el futuro planeamos incorporar métodos no lineales, utilizando técnicas de aprendizaje profundo basadas en redes neuronales para mejorar nuestro modelo, "Dijo Ahalawat.