Los investigadores de Rice University y Baylor College of Medicine utilizaron simulaciones por computadora para estudiar el proceso por el cual la hemaglutinina ayuda a los virus a invadir e infectar las células. Los investigadores creen que el dominio del tallo de la proteína se despliega y se pliega en una configuración diferente cuando se activa, pero se detiene para liberar un péptido de fusión oculto que une el virus a la célula diana. Haga clic en la imagen para una versión más grande. Crédito:Xingcheng Lin
Hay un problema en la oscilación de una proteína que transmite el virus de la gripe. Los investigadores de Rice University y Baylor College of Medicine creen que este mecanismo puede ser un objetivo útil para evitar que el virus infecte las células.
En un artículo publicado en Proceedings of the National Academy of Sciences, el equipo de Rice-Baylor dirigido por el biofísico José Onuchic y los bioquímicos Jianpeng Ma y Qinghua Wang profundiza en un complejo de glicoproteínas que comenzó a definir en un artículo de 2014.
Esa proteína hemaglutinina, se asienta sobre la superficie de los virus de la gripe y los ayuda a adherirse y transportarse a través de las membranas protectoras de las células diana.
El documento comienza a definir el mecanismo que permite que la proteína se despliegue y se repliegue en un abrir y cerrar de ojos. cambiando su forma para exponer un péptido que une el virus a una célula y comienza la infección. Los investigadores creen que los medicamentos terapéuticos pueden utilizar este mecanismo para apagar el virus.
"Esta proteína comienza en un estado plegado y pasa por una transformación global, replegarse en un estado completamente diferente, "dijo Onuchic, codirector del Centro de Rice de Física Biológica Teórica (CTBP). "Pero hay una pequeña parte en el centro que la evolución ha conservado".
Ese único residuo de aminoácido conservado es el problema que hace que la proteína se detenga en el proceso de replegamiento. Permite que un péptido de fusión enterrado en el interior se una a la célula diana y comience a infectarla. Sin pausa el plegado sería demasiado rápido para que se realizara la encuadernación.
El autor principal e investigador postdoctoral de Rice, Xingcheng Lin, modeló esa parte de la proteína, el bucle B del dominio HA2. HA2 se encuentra debajo de otro dominio, una gorra conocida como HA1 que muta para escapar de las defensas pasadas. Lin explicó que HA1 es un objetivo común para los medicamentos contra la gripe porque el dominio cap expuesto es más accesible que el dominio HA2 protegido.
El problema es que HA1 muta constantemente para resistir las drogas, él dijo. Eso influye en la eficacia de las vacunas contra la influenza cada año. Lin y Onuchic dijeron que HA2 presenta un mejor objetivo para las drogas porque el mecanismo está altamente conservado por la evolución.
"Si un fármaco se dirige a HA2, el dominio no puede escapar haciendo mutaciones porque las mutaciones mismas lo harían no funcional, ", Dijo Lin." Ese tipo de medicamento podría convertirse en una vacuna universal ".
HA2 es una estructura trimérica que, cuando se activa por condiciones ácidas en el medio ambiente cerca de una célula objetivo, se transforma de un bucle aleatorio a una bobina enrollada. Incluso con la pausa se despliega y se despliega en una fracción de segundo, demasiado rápido para que los microscopios lo vean. Pero una simulación por computadora del proceso puede ralentizarse.
Eso pasa a ser una especialidad del CTBP, que utiliza programas que analizan el panorama energético de las proteínas para predecir cómo se plegarán. Onuchic y sus colegas son pioneros en la teoría de que las proteínas de plegamiento siguen un orden proceso "canalizado" que depende de la energía intrínseca de cada átomo de la cadena, cada uno de los cuales busca constantemente su estado de energía más bajo. Si se pueden identificar todas las "perlas" atómicas, es posible simular el complejo proceso de plegado.
Los investigadores de Rice a menudo utilizan modelos de proteínas de grano grueso, un subconjunto de átomos que representan el todo, para predecir cómo se doblarán. El nuevo estudio fue mucho más ambicioso y se propuso predecir el complejo despliegue y replegamiento utilizando no solo todos los átomos de la cadena, sino también todos los átomos de su entorno líquido. Dijo Onuchic.
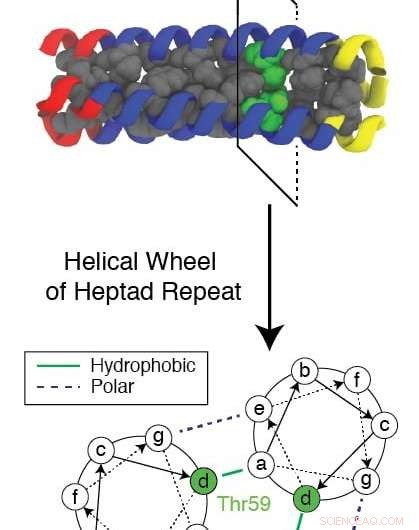
Un residuo conservado en la evolución conocido como Thr59 interrumpe el patrón repetitivo formado por una proteína trimérica a medida que se pliega mientras ayuda a que el virus de la gripe infecte una célula. Investigadores de Rice University y Baylor College of Medicine utilizaron una compleja simulación por computadora para estudiar el mecanismo y buscar nuevos objetivos para los medicamentos para detener la gripe. Crédito:Xingcheng Lin
Lin modeló 40 microsegundos (millonésimas de segundo) de la transición del dominio HA2 que representa el proceso completo, que tarda 1,4 milisegundos (milésimas de segundo) en completarse. Incluso ese proceso abreviado tomó dos años de tiempo de computadora para generar resultados, él dijo.
"El dominio simulado es aproximadamente 3, 000 átomos, pero cuando el ambiente, incluyendo agua, se contabiliza, la simulación total incorpora alrededor de 100, 000 átomos, ", Dijo Onuchic." Sigue siendo una enorme simulación que requirió técnicas de vanguardia ".
Las teorías anteriores basadas en imágenes cristalográficas de las proteínas del antes y el después plantean la idea de un dominio cargado por resorte que parecía adherirse a la célula diana después de la extracción de la tapa. Onuchic dijo que el modelo completo de HA2 admite un mecanismo diferente.
"Descubrimos que hay una gran cantidad de energía que hace que el estado final de HA2 sea mucho más estable que el estado inicial, ", dijo." Pero con el mecanismo de resorte, la mayor parte de la energía ya se habrá desperdiciado cuando se forme la bobina enrollada y se una a las membranas celulares y virales. No dejaría energía para juntar las membranas.
"Por eso decidimos hacer un cálculo completo del sistema:todos los átomos de la proteína y toda el agua, "Dijo Onuchic." Fue un esfuerzo gigantesco ".
El residuo hidrófilo conservado (que atrae el agua), conocido como Thr59, es de particular interés para los investigadores no solo por la forma en que interrumpe el plegamiento y permite que el virus ataque, pero también porque tiene un gemelo.
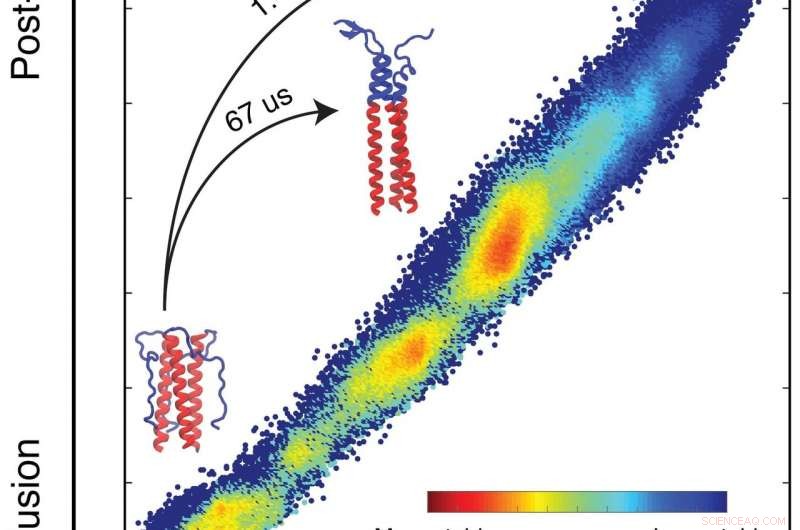
Una simulación realizada por biofísicos de la Universidad de Rice detalló el perfil de energía libre que dicta cómo una proteína que ayuda al virus de la gripe a infectar las células lleva a cabo su misión. Las simulaciones predicen cómo se plegará una proteína en función de las energías intrínsecas de cada átomo del sistema. Las proteínas forman bucles y espirales mientras buscan lo más bajo, estados de energía más estables (azul). En el dominio que estudiaron los investigadores, encontraron un problema que ralentiza el proceso de plegado que permite que se produzca la unión a la célula diana y también presenta una oportunidad para que las nuevas vacunas ataquen la gripe. Haga clic en la imagen para una versión más grande. Crédito:Xingcheng Lin
"En el árbol evolutivo completo, estos virus se dividen en dos grupos, y la diferencia parece ser este residuo, "Dijo Onuchic." Se dividieron 1, Hace 500 años y de alguna manera, después de esta separación, están totalmente conservados. No han podido cambiar ese residuo pase lo que pase, y creemos que eso hace que este residuo sea importante ".
La investigación actual se centró en el grupo que incorpora Thr59 y causa la cepa H3N2 responsable de la gripe de Hong Kong, Dijo Lin. El otro residuo, Met59, aparece en la cepa H1N1 que causó la gripe española.
"Todavía tenemos un largo camino por recorrer para comprender la proteína completa, ", dijo." Aquí, solo estudiamos un dominio de una proteína, y hay varios otros que son muy importantes para su función ".
"Pero lo que Xingcheng ya ha hecho es un tour de force computacional, "Agregó Onuchic." Mostró cómo este residuo en particular rompe la simetría helicoidal del dominio y lo hace lo suficientemente inestable como para que el péptido tenga tiempo de agarrar las membranas ".