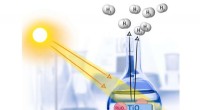
Figura 1:Ángulo diedro (el ángulo formado por el plano creado por los átomos A, B, y C, y el plano creado por los átomos B, C, y D). Crédito:Fujitsu
Fujitsu Laboratories anunció hoy el desarrollo de tecnología de simulación molecular para el descubrimiento de fármacos que puede estimar con precisión la afinidad de unión. que representa el grado en que las proteínas que pueden causar enfermedades (proteínas diana) se unen a sustancias químicas que podrían convertirse en fármacos candidatos. En el proceso de descubrimiento de fármacos, existe una demanda de una predicción precisa de la afinidad de unión entre las proteínas diana y las sustancias químicas, que ofrece una estimación aproximada de la eficacia de un fármaco. La tecnología de simulación molecular se ha utilizado ampliamente en el pasado como método para predecir la afinidad de unión, calcular las fuerzas aproximadas que surgen entre átomos en moléculas usando la mecánica newtoniana. El problema con este método, sin embargo, Queda por el bajo grado de precisión de su estimación de los parámetros más importantes:el grado de torsión en los sitios de unión. Esto significa que la precisión de su estimación de la afinidad de unión global también es mala.
Ahora, Fujitsu Laboratories ha desarrollado una tecnología de simulación molecular que estima el grado de torsión de una sustancia química, que está directamente conectado a la afinidad de unión predicha. La nueva tecnología no solo tiene en cuenta el lugar de unión donde se producirá la torsión, pero también el impacto de los átomos vecinos. Fujitsu Laboratories evaluó esta tecnología para 190 tipos de sustancias químicas, comparar los resultados con los resultados correctos obtenidos a partir del cálculo de los primeros principios y luego evaluar la tasa de error. Al hacerlo, pudo confirmar que la tasa de error en la estimación del grado de torsión era, de media, una décima parte de la de la tecnología anterior. Se prevé que el uso de esta nueva tecnología en el descubrimiento de fármacos basados en TI, con su capacidad para estimar con precisión la afinidad de unión de proteínas y sustancias químicas específicas, ofrece el potencial para esfuerzos innovadores de descubrimiento de nuevos fármacos que no se podrían lograr con enfoques anteriores.
El descubrimiento de nuevos medicamentos requiere importantes gastos y plazos que pueden medirse en décadas, conduciendo a una búsqueda global de nuevos métodos de descubrimiento de drogas. Uno de los métodos que ha recibido un interés considerable es el descubrimiento de fármacos basado en tecnologías de la información, un nuevo método de descubrimiento de fármacos mediante ordenadores que permite crear sustancias químicas candidatas a fármacos novedosos con una alta probabilidad de éxito. El descubrimiento de fármacos basado en tecnologías de la información se ha convertido en un punto focal de expectativas como tecnología innovadora para la creación de nuevos fármacos. porque a diferencia de los métodos anteriores de prueba y error, en el que las sustancias químicas se crean y prueban repetidamente, este enfoque permite diseñar virtualmente sustancias químicas y estimar sus efectos.
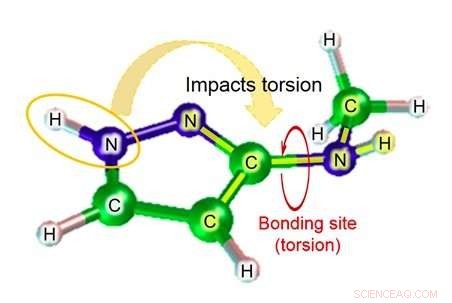
Figura 2:Ejemplo de estructura molecular:3- (metilamino) pirazol. Crédito:Fujitsu
Los efectos de una sustancia química como fármaco se expresan cuando la sustancia química se une a una proteína objetivo. Cuando la sustancia química se une a la proteína objetivo, puede cambiar su forma de acuerdo con la de la proteína objetivo. El grado de deformación, a saber, los parámetros que indican la extensión de este cambio de forma, está directamente relacionado con la afinidad de unión de la sustancia y la proteína, y da una idea aproximada de su efecto como droga. Dado este, Existe una fuerte demanda de la capacidad de predecir con precisión este valor. Para calcular el grado de deformación de una sustancia química, existen métodos basados en la mecánica cuántica y métodos basados en la mecánica newtoniana. El cálculo de los primeros principios basado en la mecánica cuántica permite cálculos extremadamente precisos, resolviendo los estados de los electrones a partir de los tipos y posiciones de los átomos involucrados. Por otra parte, sin embargo, la capacidad de los primeros principios para realizar cálculos rigurosos conduce necesariamente a una enorme cantidad de tiempo necesario para completar los cálculos. Para simular el grado de deformación de numerosas sustancias químicas, el tiempo requerido es del orden de años, haciendo este método poco práctico. Por otra parte, Los cálculos aproximados basados en simulaciones moleculares son extremadamente rápidos, utilizando la mecánica newtoniana para calcular las fuerzas entre los átomos dentro de las moléculas, e incluso puede manejar moléculas grandes como proteínas con bastante facilidad. Como consecuencia, este método es muy utilizado. Con la mecánica newtoniana, las fuerzas entre los átomos se expresan de la siguiente manera:
Entre estos, cuando una sustancia química se une a una proteína objetivo, el grado de torsión de la unión representa el importante grado de deformación. Con la tecnología existente, sin embargo, la precisión de la estimación del parámetro del ángulo diedro (figura 1), que es necesario para calcular el grado de torsión del enlace, es bastante bajo, resultando en el problema de baja precisión en la estimación de la afinidad del enlace en la simulación.
Fujitsu Laboratories lleva más de diez años desarrollando tecnología de simulación molecular. Ahora, utilizando el conocimiento obtenido a través de esfuerzos previos, Fujitsu Laboratories ha desarrollado una tecnología de simulación molecular que puede estimar el parámetro del ángulo diedro teniendo en cuenta el impacto de los átomos cerca del enlace. La tecnología existente estima el parámetro del ángulo diedro basándose en un total de cuatro átomos:los dos átomos en el enlace relevante, y los otros átomos a los que estaba unido cada uno de esos átomos. Dependiendo de la estructura de la molécula, sin embargo, hay casos en los que átomos más allá de esos cuatro podrían tener un impacto significativo, y en esos casos, el margen de error de la estimación podría ser bastante grande. Con esta tecnología, Fujitsu Laboratories ha creado una base de datos de fórmulas de estimación para patrones de estructura parcial donde el impacto de los átomos más alejados del sitio de enlace podría ser significativo. así como por el grado de torsión de sustancias químicas que se esperaría en ese caso. Usando la fórmula de estimación relevante para encontrar el grado de torsión (figura 2) en el caso de moléculas correspondientes a la base de datos para estructuras parciales, se ha hecho posible incluso realizar estimaciones muy precisas de la torsión molecular, que anteriormente era difícil de calcular con precisión.
Cuando Fujitsu Laboratories integró esta tecnología en el software que había desarrollado para generar parámetros sofisticados para las fuerzas entre átomos (FF-FOM), pudo confirmar que los resultados se ajustaban a cálculos precisos.

Figura 3:Evaluación del rendimiento de los valores de los parámetros de ángulo diedro utilizando 190 tipos de estructuras de compuestos químicos. Crédito:Fujitsu
Cuando Fujitsu Laboratories evaluó la diferencia entre los resultados de esta tecnología y los resultados de un cálculo a partir de primeros principios para la estimación del grado de torsión con 190 tipos de sustancias químicas, era menos de una décima parte de la tecnología anterior, de media, 0,6 kcal / mol por debajo de las fluctuaciones térmicas de la temperatura ambiente, confirmando que la nueva tecnología es práctica. Debido a que puede estimar con precisión la afinidad de unión de las proteínas y sustancias químicas objetivo, Se espera que el uso de esta tecnología conduzca a la creación de nuevos fármacos revolucionarios mediante su uso en el descubrimiento de fármacos basados en tecnologías de la información.