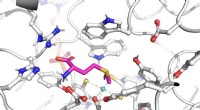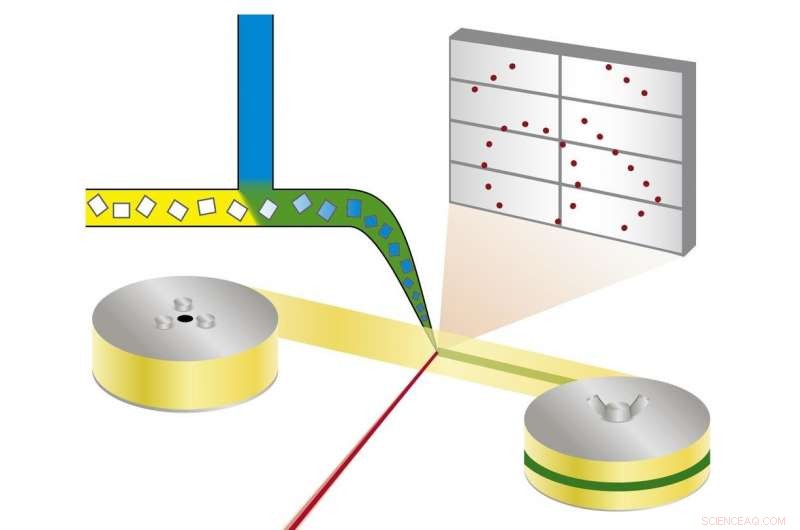
Principio de la cristalografía de sincrotrón en serie de mezcla y difusión:los cristales de proteína se mezclan con una solución de un candidato a fármaco y se radiografían en una cinta que atraviesa el haz de rayos X. Crédito:Beyerlein et al., IUCrJ
Los científicos de DESY han desarrollado un nuevo método que permite la detección rápida y automatizada de candidatos a fármacos prometedores. Esta novedosa técnica, llamada cristalografía de sincrotrón en serie mixta y difusa, Puede visualizar la interacción de posibles dianas farmacológicas con candidatos farmacológicos u otras moléculas. El concepto tiene el potencial de llevar el diseño de fármacos basado en estructura y fragmentos a un nuevo nivel, como escriben los investigadores en el Revista de la Unión Internacional de Cristalografía ( IUCrJ ).
Muchas proteínas del cuerpo son posibles dianas farmacológicas. Las moléculas farmacéuticas con la forma correcta pueden unirse a estas proteínas y activar o desactivar su función. Por ejemplo, para combatir ciertas formas de leucemia, el medicamento contra el cáncer Imatinib inhibe una variante hiperactiva de la enzima tirosina-quinasa, una proteína responsable de activar muchas otras proteínas. Imatinib bloquea el sitio activo de esta tirosina quinasa. Para lograr esto, la molécula del fármaco debe encajar con precisión en el sitio activo como una llave en una cerradura. Basado en el conocimiento de la estructura espacial de la enzima diana, Imatinib se diseñó a medida para este fin.
"Esta estrategia se denomina diseño de fármacos basado en la estructura y hoy en día se utiliza como método estándar en el desarrollo de fármacos farmacéuticos, "explica el primer autor Kenneth Beyerlein del Center for Free-Electron Laser Science (CFEL), una cooperación de DESY, la Universidad de Hamburgo y la Sociedad Alemana Max Planck. "Sin embargo, en realidad, apuntar a las proteínas es mucho más complejo que colocar una llave en una cerradura. Por lo tanto, muchas moléculas farmacéuticas potenciales o fragmentos de tales moléculas deben probarse, que suele ser un procedimiento largo y complicado ". Además, tanto los biólogos como los farmacólogos están interesados en el funcionamiento preciso de los agentes naturales que se unen a las proteínas, para comprender mejor la maquinaria de la vida.
El sistema desarrollado por el equipo de Beyerlein y su colega de DESY Dominik Oberthür, también de CFEL, ofrece una nueva forma de perseguir este objetivo:mezcla proteínas microcristalinas con moléculas específicas llamadas ligandos que pueden ser candidatos a fármacos o agentes naturales justo antes de sondear los cristales con rayos X para revelar la estructura espacial detallada del complejo proteína-ligando resultante o el ausencia de tal complejo si un ligando potencial no se une a la proteína.
Para analizar la estructura espacial de una proteína, los científicos suelen utilizar la cristalografía de rayos X. Para esta técnica, primero hay que cultivar un cristal a partir de la proteína. Luego, los investigadores toman instantáneas de rayos X de todos los lados del cristal que debe enfriarse a temperaturas ultrabajas para reducir el daño de la intensa radiación. Los rayos X producen un patrón de difracción característico a partir del cual se puede calcular la estructura interna del cristal y, por lo tanto, la estructura espacial de la proteína. Para investigar una proteína con un ligando, se debe hacer crecer un nuevo cristal a partir de una solución de proteína y ligando o el cristal debe empaparse con el ligando. Incluso con el uso de robótica para automatizar todos los pasos de este proceso, la necesidad de montar cristales individuales para cada nuevo conjunto de datos se ha convertido en el paso que limita la velocidad en el cribado de grandes bibliotecas de compuestos.
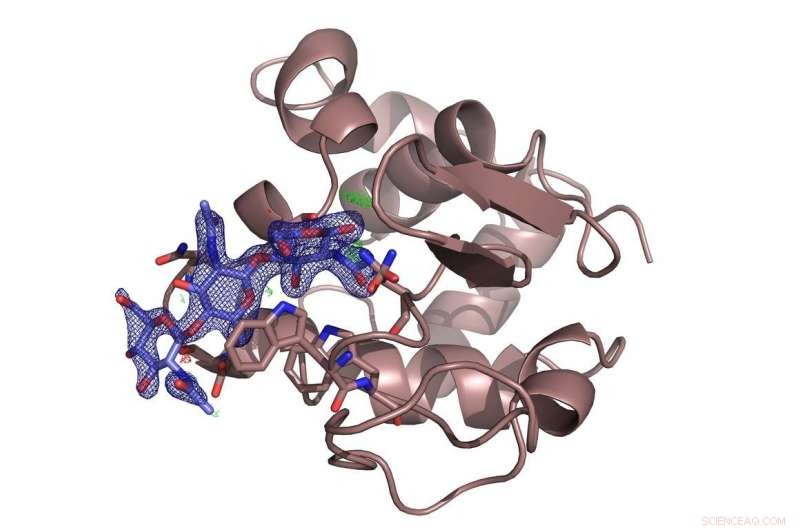
La enzima lisozima (marrón) con el azúcar inhibidor quitotriosa (azul) unida a ella. La investigación resolvió una controversia sobre el sitio de unión preferido de la molécula de azúcar. Crédito:DESY, Dominik Oberthür
La nueva técnica sigue un enfoque diferente. "Estamos usando microcristales que tienen dos ventajas:por lo general, son mucho más fáciles de producir que los cristales grandes, y son lo suficientemente pequeños como para que un fármaco potencial en una solución pueda difundirse a través del cristal y unirse a todas las moléculas de proteína en unos pocos milisegundos, "explica Oberthür. El sistema desarrollado por el equipo de Oberthür y Beyerlein dispensa una corriente de microcristales en un líquido portador en una cinta delgada. Como una cinta transportadora, la cinta transporta los cristales a través del haz de rayos X, que se corta en breves destellos por una persiana giratoria. En lugar de girar un gran cristal en el haz de rayos X, Muchos microcristales en orientación aleatoria se someten a rayos X en serie y los patrones de difracción de cada disparo se combinan posteriormente para formar un conjunto de datos completo. siguiendo el concepto de cristalografía en serie que se desarrolló por primera vez en láseres de rayos X de electrones libres (XFEL).
A través de una segunda válvula en el sistema, Se añade una solución de un candidato a fármaco o ligando natural. El punto donde los dos líquidos se mezclan se puede ajustar para crear un retraso definido antes de investigar la estructura. Esta configuración no requiere crioenfriamiento de cristales, por lo tanto, la interacción proteína-fármaco se puede observar a temperaturas fisiológicas, o cualquier otra temperatura deseada. De esta manera, incluso se puede investigar la dinámica de unión. "Podemos difundir productos químicos en los cristales de proteínas sobre la marcha y ver cómo se produce la unión, "explica Oberthür." No es necesario encontrar nuevas condiciones de crecimiento para cada inhibidor y no es necesario intercambiar los cristales manualmente, todo el proceso se puede automatizar ".
El equipo probó el nuevo sistema en la fuente de rayos X de alto brillo de DESY, PETRA III, con la conocida proteína lisozima y una molécula de azúcar. quitotriosa, que inhibe la enzima. Los microcristales de lisozima utilizados aquí tenían solo de seis a ocho micrómetros de diámetro. La configuración en la estación de medición P11 reveló la estructura espacial del inhibidor mezclado unido a la lisozima en detalle. Y aunque la estructura de la lisozima fue la primera estructura enzimática revelada por cristalografía de rayos X hace 50 años, el nuevo método aún podría revelar nuevos detalles sobre el modo de unión de la quitotriosa a la lisozima, resolviendo una controversia sobre el sitio de unión preferido de la molécula de azúcar.
Si bien la prueba de principio aún requería algo de tiempo, Los avances rutinarios y posteriores en la tecnología de rayos X y detectores acelerarán considerablemente el procedimiento. También, utilizando todo el espectro del haz de rayos X de la fuente de luz del sincrotrón en lugar de un solo "color" de la misma, puede reducir el tiempo de exposición para imágenes de difracción individuales a 100 picosegundos, o 0,1 mil millonésimas de segundo. Solo 50 de estas imágenes son suficientes para determinar la estructura, como se mostró recientemente.
"Estamos desarrollando formas de resolver la estructura de las proteínas unidas para el descubrimiento de fármacos de alto rendimiento, ", explicó Beyerlein. Como las fuentes de luz de sincrotrón son más accesibles que los láseres de rayos X, los investigadores imaginan utilizar este método para la detección sistemática a través de bibliotecas de posibles inhibidores y fragmentos de fármacos. "Hacer esto de forma automática y mucho más rápido que con los enfoques convencionales sería un gran paso adelante en el diseño de fármacos basados en la estructura, "dice Beyerlein.