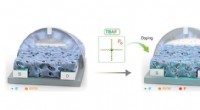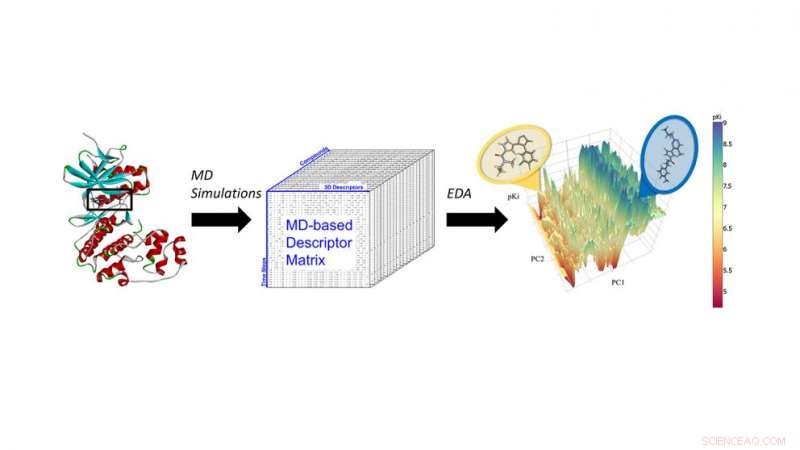
Simulaciones de dinámica molecular (MD) de inhibidores de ERK2 para extraer descriptores de MD para el análisis quiminformático de próxima generación y el aprendizaje automático. Crédito:Universidad Estatal de Carolina del Norte
Investigadores de la Universidad Estatal de Carolina del Norte han demostrado que las simulaciones de dinámica molecular y las técnicas de aprendizaje automático podrían integrarse para crear modelos de predicción por computadora más precisos. Estos modelos "hiper-predictivos" podrían usarse para predecir rápidamente qué nuevos compuestos químicos podrían ser candidatos a fármacos prometedores.
El desarrollo de fármacos es un proceso costoso y que requiere mucho tiempo. Para reducir el número de compuestos químicos que podrían ser candidatos potenciales a fármacos, Los científicos utilizan modelos informáticos que pueden predecir cómo un compuesto químico en particular podría interactuar con un objetivo biológico de interés, por ejemplo, una proteína clave que podría estar involucrada en el proceso de una enfermedad. Tradicionalmente, esto se hace a través del modelado cuantitativo de la relación estructura-actividad (QSAR) y acoplamiento molecular, que se basan en información 2 y 3-D sobre esos productos químicos.
Denis Fourches, profesor asistente de química computacional, quería mejorar la precisión de estos modelos QSAR. "Cuando examina un conjunto de 30 millones de compuestos, no necesariamente necesita una confiabilidad muy alta con su modelo, solo está obteniendo una idea aproximada sobre el 5 o 10 por ciento superior de esa biblioteca virtual. Pero si está intentando reducir un campo de 200 análogos a 10, que es más común en el desarrollo de fármacos, su técnica de modelado debe ser extremadamente precisa. Las técnicas actuales definitivamente no son lo suficientemente confiables ".
Fourches y Jeremy Ash, un estudiante de posgrado en bioinformática, decidió incorporar los resultados de los cálculos de dinámica molecular (simulaciones de todos los átomos de cómo un compuesto en particular se mueve en el bolsillo de unión de una proteína) en modelos de predicción basados en el aprendizaje automático.
"La mayoría de los modelos solo utilizan estructuras bidimensionales de moléculas, "Dice Fourches". Pero en realidad, Los productos químicos son objetos tridimensionales complejos que se mueven, vibran y tienen interacciones intermoleculares dinámicas con la proteína una vez acoplada en su sitio de unión. No puedes ver eso si solo miras la estructura 2-D o 3-D de una molécula dada ".
En un estudio de prueba de concepto, Fourches y Ash observaron la quinasa ERK2, una enzima asociada con varios tipos de cáncer, y un grupo de 87 inhibidores conocidos de ERK2. que van desde muy activos a inactivos. Ejecutaron simulaciones independientes de dinámica molecular (MD) para cada uno de esos 87 compuestos y calcularon información crítica sobre la flexibilidad de cada compuesto una vez en el bolsillo ERK2. Luego analizaron los descriptores de MD utilizando técnicas de quimioinformática y aprendizaje automático. Los descriptores de MD pudieron distinguir con precisión los inhibidores activos de ERK2 de los débilmente activos e inactivos, que no era el caso cuando los modelos usaban solo información estructural 2-D y 3-D.
"Ya teníamos datos sobre estas 87 moléculas y su actividad en ERK2, "Dice Fourches." Así que probamos para ver si nuestro modelo era capaz de encontrar de manera confiable los compuestos más activos. En efecto, distinguió con precisión entre inhibidores de ERK2 fuertes y débiles, y debido a que los descriptores MD codificaron las interacciones que esos compuestos crean en el bolsillo de ERK2, también nos dio más información sobre por qué los inhibidores fuertes funcionaron bien.
"Antes de que los avances informáticos nos permitieran simular este tipo de datos, Nos habría llevado seis meses simular una sola molécula en el bolsillo de ERK2. Gracias a la aceleración de la GPU, ahora solo lleva tres horas. Eso es un cambio de juego. Tengo la esperanza de que la incorporación de datos extraídos de la dinámica molecular en los modelos QSAR permitirá una nueva generación de modelos hiperpredecibles que ayudarán a traer novedades, medicamentos eficaces en el mercado incluso más rápido. Es la inteligencia artificial trabajando para que descubramos las drogas del mañana ".