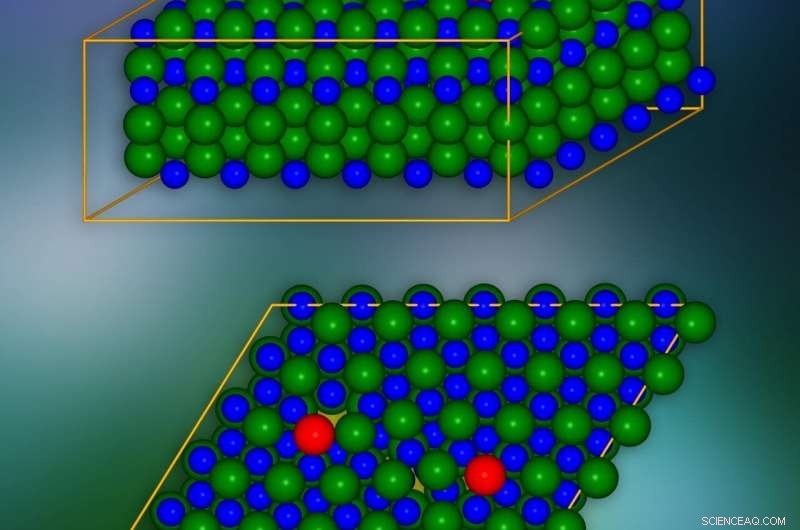
Modelo de cristal de carburo de silicio con dislocaciones de borde introducidas en lugares marcados en rojo. Un solo plano cristalográfico se presenta en la parte inferior. Los lugares donde las cargas eléctricas pueden 'filtrarse' a las capas vecinas están marcados en amarillo. Crédito:IFJ PAN
Imperfecciones de la estructura cristalina, especialmente dislocaciones de borde de naturaleza alargada, modificar profundamente las propiedades básicas de todo el material y, en consecuencia, limitar drásticamente sus aplicaciones. Usando carburo de silicio como ejemplo, físicos de Cracovia y Varsovia han demostrado que incluso esos defectos computacionalmente exigentes se pueden examinar con éxito con precisión atómica por medio de un ingeniosamente construido, pequeño en tamaño, modelo.
Las matemáticas aman la perfección. Desafortunadamente, la perfección no ama la realidad física. Los teóricos que modelan cristales han intentado durante mucho tiempo incluir defectos en estructuras cristalinas reales y predecir su impacto en las propiedades físicas de los materiales. Los modelos, basado en los resultados de varios experimentos, han descrito cambios en las propiedades básicas de un material sin explicar las causas y efectos reales de los fenómenos que ocurren.
Un nuevo modelo de carburo de silicio (SiC), construido por físicos del Instituto de Física Nuclear de la Academia Polaca de Ciencias (IFJ PAN) en Cracovia, les ha permitido demostrar que ahora es posible estudiar cristales ab initio con defectos tan complejos como las dislocaciones de los bordes y explicar sus características mediante procesos que ocurren a escala atómica. Este resultado espectacular, presentado recientemente en la conferencia Multiscale Phenomena in Molecular Matter 2019 en Cracovia, fue logrado por los físicos del PAN de la FIP en cooperación con el Instituto de Investigación Tecnológica Fundamental de la Academia de Ciencias de Polonia y el Instituto de Física de Alta Presión de la Academia de Ciencias de Polonia, ambos ubicados en Varsovia.
“Intentamos encontrar los mecanismos responsables a nivel atómico de reducir el voltaje de ruptura en los cristales de carburo de silicio. Nuestros cálculos ab initio conducen a una comprensión cualitativa del problema y contribuyen a explicar los detalles de este fenómeno, "dice el Dr. Jan Lazewski, profesor de la FIP PAN.
Los cálculos ab initio tienen ahora una larga historia relacionada con el Premio Nobel de Walter Kohn y John Pople en 1998 (sin embargo, las simulaciones de defectos cristalinos lineales solo se han introducido recientemente). Este término se utiliza para describir los cálculos realizados utilizando ecuaciones de mecánica cuántica, apoyado únicamente por el conocimiento sobre la estructura del átomo y la simetría de los cristales. No hay información directa de experimentos en tales modelos, lo que significa que también se pueden utilizar para analizar materiales que nunca antes se han estudiado o incluso sintetizado. Debido a una complicación relativamente importante del problema, hasta ahora los cálculos ab initio funcionaron, a lo sumo, en el caso de defectos puntuales, relacionados con las vacantes (átomos faltantes o huecos en la estructura cristalina), así como con las mezclas introducidas en el cristal.
No en vano los investigadores de Cracovia utilizaron carburo de silicio. Las propiedades de este semiconductor son tan interesantes que en el pasado incluso se consideró un sucesor del silicio. Su banda prohibida (la barrera que tiene que superar la carga para pasar de la banda de valencia a la banda de conducción y conducir la corriente) es casi tres veces mayor que en el silicio, la densidad de corriente de conducción permisible:el doble, la capacidad de disipar el calor, más de tres veces mayor, y la frecuencia de corte de la operación del cristal hasta seis veces mayor. Además, Los sistemas de carburo de silicio pueden operar a temperaturas de hasta 650 grados Celsius, mientras que los sistemas de silicio ya empiezan a tener problemas a 120 grados centígrados. El SiC también tiene un alto punto de fusión, es difícil, resistente al ácido y la radiación. Sus desventajas incluyen sobre todo el precio:mientras que las obleas de silicio de dos pulgadas cuestan solo unos pocos dólares, el valor de obleas de carburo de silicio similares asciende a miles. Los cristales de carburo de silicio de baja calidad son un material abrasivo popular, También se utiliza en chalecos antibalas y en los discos de freno de los coches más caros del mundo. como Lamborghini o Bugatti. Los cristales de alta calidad se utilizan para producir espejos para telescopios y en dispositivos de alto voltaje con alta resistencia a la temperatura.
A nivel atómico, Los cristales de carburo de silicio se componen de muchas capas planas dispuestas una encima de la otra. Cada capa se asemeja a un panal:consta de celdas hexagonales en las que las moléculas de carburo de silicio se ubican verticalmente en las esquinas. Cada dos capas adyacentes se pueden combinar de tres formas. Los 'sándwiches' multicapa con diferentes diseños crean los llamados politipos, de los cuales existen más de 250 en el caso del carburo de silicio. El grupo de IFJ PAN utilizó el polimorfo 4H-SiC.
"Al modelar estas estructuras, uno de los principales problemas es la complejidad computacional. Un modelo de cristal puro, desprovisto de aditivos o dislocaciones, se caracteriza por una alta simetría y se puede calcular incluso en unos pocos minutos. Para realizar un cálculo de un material con dislocación, necesitamos meses trabajando en una computadora de alta potencia, "enfatiza el Dr. Pawel Jochym, profesor de la FIP PAN.
Los problemas con las dislocaciones de los bordes se deben a la escala de su influencia en la estructura cristalina del material. Como una ilustracion, pueden compararse con el problema de disfrazar un hueco en una hilera de baldosas en un suelo. El espacio se puede 'camuflar' moviendo los mosaicos de las filas adyacentes, pero el defecto siempre será visible. Las dislocaciones de borde que resultan de la falta de longitudes completas o regiones de átomos / moléculas en capas de cristal individuales actúan de manera similar, afectando las posiciones de átomos y moléculas en muchas capas adyacentes. Y dado que las dislocaciones pueden extenderse a largas distancias, en la práctica, las perturbaciones causadas por ellos incluyen todo el cristal.
Los fenómenos más interesantes tienen lugar en el núcleo de la dislocación, es decir, en las proximidades del borde de la capa dañada de la red de cristal. Para eliminar los efectos a largo plazo causados por una sola dislocación, y así reducir significativamente el número de átomos en consideración, Se empleó un truco:se introdujo una segunda dislocación del efecto contrario. De este modo, se compensó el impacto de la primera dislocación en distancias más largas.
El modelo de cristal de SiC constaba de unos 400 átomos. Las simulaciones mostraron que en las capas de cristales, a lo largo del borde del núcleo del defecto, Los 'túneles' aparecen en forma de canales con densidad de carga reducida. Disminuyen la barrera de potencial localmente y hacen que las cargas eléctricas se "filtren" de la banda de valencia. Además, en la brecha prohibida, que en el aislante garantiza una falta de conductividad eléctrica, Aparecen condiciones que reducen su amplitud y efectividad para limitar el flujo de carga. Se demostró que estos estados se originan a partir de átomos ubicados en el núcleo de dislocación.
"La situación se puede comparar con una profunda, barranco empinado que una ardilla intenta cruzar. Si el fondo del barranco está vacío, la ardilla no llegará al otro lado. Sin embargo, si hay varios árboles en la parte inferior que son lo suficientemente altos, la ardilla puede saltar sobre sus copas al otro lado del barranco. En el cristal que modelamos las ardillas son las cargas eléctricas, la banda de valencia es un borde del barranco, la banda de conducción es la otra, y los árboles son los estados antes mencionados asociados con los átomos del núcleo de dislocación, "dice el profesor Lazewski.
Ahora que se conocen los mecanismos responsables de bajar el umbral de la barrera energética a nivel atómico, hay un gran margen para la experimentación. El mecanismo propuesto deberá ser verificado para poder utilizarlo para limitar la influencia negativa de los defectos probados. Afortunadamente, ya existen posibilidades técnicas para ello.
"El futuro verificará si nuestras ideas se confirmarán en su totalidad. Sin embargo, confiamos en el destino de nuestro modelo y el enfoque presentado para simular las dislocaciones de los bordes. Ya sabemos que el modelo ab initio ha demostrado su valía frente a ciertos datos experimentales, "concluye el profesor Jochym.