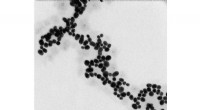
El aprendizaje automático predice la estructura y la dinámica de las nanopartículas. Las nanoestructuras como estas nanopartículas de oro cubiertas de tiol ahora se pueden estudiar mediante el nuevo método de aprendizaje automático desarrollado en la Universidad de Jyväskylä. El método puede predecir la energía potencial de una estructura dada de manera confiable. Crédito:Antti Pihlajamäki / Universidad de Jyväskylä
Investigadores del Centro de Nanociencia y de la Facultad de Tecnología de la Información de la Universidad de Jyväskylä en Finlandia han demostrado que los nuevos métodos de aprendizaje automático a distancia desarrollados en la Universidad de Jyväskylä son capaces de predecir las estructuras y la dinámica atómica de las nanopartículas de forma fiable. Los nuevos métodos son significativamente más rápidos que los métodos de simulación tradicionales utilizados para la investigación de nanopartículas y facilitarán exploraciones más eficientes de las reacciones de partículas a partículas y la funcionalidad de las partículas en su entorno. El estudio se publicó en un número especial dedicado al aprendizaje automático en el Revista de química física el 15 de mayo 2020.
Los nuevos métodos se aplicaron a nanopartículas metálicas estabilizadas con ligando, que se han estudiado durante mucho tiempo en el Centro de Nanociencia de la Universidad de Jyväskylä. El año pasado, los investigadores publicaron un método que puede predecir con éxito los sitios de unión de las moléculas del ligando estabilizador en la superficie de las nanopartículas. Ahora, se creó una nueva herramienta que puede predecir de manera confiable la energía potencial basada en la estructura atómica de la partícula, sin la necesidad de utilizar cálculos de estructura electrónica numéricamente pesados. La herramienta facilita las simulaciones de Monte Carlo de la dinámica del átomo de las partículas a temperaturas elevadas.
La energía potencial de un sistema es una cantidad fundamental en la nanociencia computacional, ya que permite evaluaciones cuantitativas de la estabilidad del sistema, tasas de reacciones químicas y fuerza de los enlaces interatómicos. Las nanopartículas metálicas estabilizadas con ligandos tienen muchos tipos de enlaces interatómicos de diferente resistencia química, y tradicionalmente, las evaluaciones de energía se han realizado utilizando la llamada teoría funcional de la densidad (DFT) que a menudo da como resultado cálculos numéricamente pesados que requieren el uso de supercomputadoras. Esto ha impedido simulaciones eficientes para comprender las funcionalidades de las nanopartículas, p.ej., como catalizadores, o interacciones con objetos biológicos como proteínas, virus o ADN. Métodos de aprendizaje automático, una vez capacitado para modelar los sistemas de manera confiable, puede acelerar las simulaciones en varios órdenes de magnitud.
El nuevo método permitió ejecutar simulaciones en una computadora portátil o de escritorio
En este trabajo los investigadores utilizaron las energías potenciales, predicho por el método de aprendizaje automático, para simular la dinámica atómica de nanopartículas de oro estabilizadas con tiol. Los resultados concuerdan bien con las simulaciones realizadas utilizando la teoría funcional de la densidad. El nuevo método permitió que las simulaciones se ejecutaran en una computadora portátil o de escritorio en una escala de tiempo de unas pocas horas, mientras que las simulaciones DFT de referencia tomaban días en una supercomputadora y usaban simultáneamente cientos o incluso miles de núcleos de computadora. La aceleración permitirá simulaciones a largo plazo de los cambios estructurales de las partículas y las reacciones partícula-partícula a temperaturas elevadas.
Los investigadores utilizaron un método de aprendizaje automático a distancia desarrollado en el grupo del profesor Tommi Kärkkäinen en Jyväskylä. Describe cada configuración atómica momentánea de una nanopartícula calculando un denominado descriptor, y compara distancias entre descriptores en un espacio numérico multidimensional. Mediante el uso de correlaciones con un conjunto de entrenamiento creado por las simulaciones DFT de referencia, la energía potencial se puede predecir. Este enfoque, utilizado ahora por primera vez en la investigación de nanopartículas, es más simple y transparente que las redes neuronales utilizadas tradicionalmente.
"Es sumamente motivador que podamos reducir la carga computacional de ejecutar simulaciones en supercomputadoras a ejecutarlas con una calidad similar en una computadora portátil o una PC doméstica, "dice el estudiante de doctorado Antti Pihlajamäki, quien es el autor principal del estudio.
"Fue una gran sorpresa que nuestros métodos de aprendizaje automático relativamente simples funcionen tan bien para nanoestructuras complicadas, "afirma el profesor Tommi Kärkkäinen.
"En la siguiente fase, Nuestro objetivo es generalizar el método para que funcione bien con nanopartículas de diferentes tamaños y composiciones químicas. Seguiremos necesitando supercomputadoras para generar suficientes datos de alta calidad para entrenar el algoritmo de aprendizaje automático, pero esperamos que en el futuro podamos pasar a utilizar estos nuevos métodos principalmente para estudios de la funcionalidad de las nanopartículas en entornos químicos complicados, "resume el profesor de la Academia Hannu Häkkinen, quien coordinó el estudio.