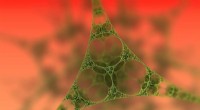
Representación esquemática de diferentes términos energéticos que contribuyen a la energía de adsorción, y diferencia de densidad de carga de 2H-P después de la adsorción sobre Cu (111) a una separación de 12,8 Angstrom. Crédito:M. Müller / TU Munich
A medida que continuamos encogiendo los componentes electrónicos, Los métodos de fabricación de arriba hacia abajo comienzan a acercarse a un límite físico a nanoescala. En lugar de seguir reduciendo este límite, una solución de interés implica el uso del autoensamblaje de abajo hacia arriba de bloques de construcción moleculares para construir dispositivos a nanoescala.
El autoensamblaje exitoso es una danza elaboradamente coreografiada, en el que las fuerzas atractivas y repulsivas dentro de las moléculas, entre cada molécula y sus vecinas, y entre las moléculas y la superficie que las sostiene, hay que tenerlo todo en cuenta. Para comprender mejor el proceso de autoensamblaje, Los investigadores de la Universidad Técnica de Munich han caracterizado las contribuciones de todos los componentes de interacción, tales como enlaces covalentes e interacciones de van der Waals entre moléculas y entre moléculas y una superficie.
"En un caso ideal, el dispositivo más pequeño posible tiene el tamaño de un solo átomo o molécula, "dijo Katharina Diller, quien trabajó como investigador postdoctoral en el grupo de Karsten Reuter en la Universidad Técnica de Munich. Reuter y sus colegas presentan su trabajo esta semana en La Revista de Física Química .
Un ejemplo es un cambio de porfirina simple, que ocupa una superficie de solo un nanómetro cuadrado. La molécula de porfina, que fue el objeto de este estudio, es incluso más pequeño que esto. Las porfirinas son un grupo de compuestos químicos anillados que incluyen, en particular, el hemo, responsable del transporte de oxígeno y dióxido de carbono en el torrente sanguíneo, y la clorofila. En aplicaciones de origen sintético, las porfirinas se estudian por sus posibles usos como sensores, tintes sensibles a la luz en células solares orgánicas, e imanes moleculares.
Los investigadores de TU Munich evaluaron las interacciones de la molécula de porfirina 2H-porfina utilizando la teoría funcional de la densidad, un método de modelado computacional de mecánica cuántica utilizado para describir las propiedades electrónicas de moléculas y materiales. Sus simulaciones se realizaron en la supercomputadora de alto rendimiento SuperMUC en Leibniz-Rechenzentrum en Garching.
Los sustratos metálicos que los investigadores eligieron para que las moléculas de porfirina se ensamblaran, las superficies de cristal único compactas de cobre y plata, se utilizan ampliamente como sustratos en la ciencia de superficies. Esto se debe a la naturaleza densamente compacta de las superficies, que permiten que las moléculas exhiban un ambiente de adsorción suave. Adicionalmente, el cobre y la plata reaccionan cada uno de manera diferente con las porhirinas:la molécula se adsorbe más fuertemente en el cobre, mientras que la plata hace un mejor trabajo al mantener intacta la estructura electrónica de la molécula, lo que permite a los investigadores monitorear una variedad de efectos en competencia para aplicaciones futuras.
En su simulación, Las moléculas de porfirina se colocaron en una placa de cobre o plata, que se repitió periódicamente para simular una superficie extendida. Después de encontrar la geometría óptima en la que las moléculas se adsorberían en la superficie, los investigadores alteraron el tamaño de la losa de metal para aumentar o disminuir la distancia entre moléculas, simulando así diferentes coberturas moleculares. La configuración computacional les dio un interruptor para encender y apagar las contribuciones de energía de las moléculas vecinas, para observar la interacción de las interacciones individuales.
Diller y Reuter, junto con sus colegas Reinhard Maurer y Moritz Müller, quién es el primer autor del artículo, encontró que las interacciones débiles de van der Waals de largo alcance producían la mayor contribución a la interacción molécula-superficie, y demostró que los métodos empleados a menudo para cuantificar las cargas electrónicas en el sistema deben utilizarse con precaución. Asombrosamente, mientras que las interacciones directas entre moléculas son insignificantes, el investigador encontró indicios de interacciones molécula-molécula mediadas por la superficie en coberturas moleculares más altas.
"El análisis de la estructura electrónica y los componentes de interacción individuales nos permite comprender mejor el autoensamblaje de la porfina adsorbida en cobre y plata, y además permite predicciones para análogos de porfirina más complejos, "Diller dijo." Estas conclusiones, sin embargo, llegar sin considerar los efectos del movimiento atómico a temperatura finita, que no estudiamos en este trabajo ".