Los tríos de átomos de tungsteno están muy influenciados en su migración a través de la naturaleza de una partícula diminuta por la forma de la partícula, según un equipo de expertos, incluyendo al Dr. Fei Gao del Laboratorio Nacional del Noroeste del Pacífico. El equipo de EE. UU. Y China realizó complejas simulaciones computacionales para determinar la energía involucrada en la migración del clúster de tungsteno. Descubrieron que el adatom de 3 a 4, o átomo de superficie, los grupos prefieren formar islas compactas. La reorientación es el mecanismo de migración dominante para el dímero, mientras que la migración neta de los racimos lager se puede lograr mediante la cizalladura del dímero, Mecanismos concertados de movimiento y rotación.
La investigación se destacó en la portada del European Physical Journal B en marzo de 2011 junto con el artículo revisado por pares:"Migración de clústeres de tungsteno en nanopartículas:un estudio del método de dímeros".
La demanda de miniaturización de dispositivos electrónicos se beneficiará de una comprensión más profunda de los materiales nanoestructurados. El tungsteno tiene propiedades únicas como alta densidad, dureza, Temperatura de fusión, elasticidad y conductividad, junto con una baja expansión térmica. Estas propiedades únicas y partículas de tamaño nanométrico se pueden utilizar para almacenar y organizar electrones para su uso por semiconductores, proporcionando a los ingenieros un material de menor resistencia y conductividad mejorada.
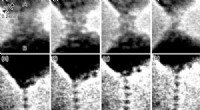
Utilizando supercomputadoras en el Laboratorio de Ciencias Moleculares Ambientales, el equipo de investigación realizó los cálculos necesarios para buscar posibles estados de transición y rutas de migración para grupos de tungsteno en nanopartículas de tungsteno, y las energías de migración correspondientes para las posibles rutas de migración de estos conglomerados.
Se ha descubierto que los grupos de tungsteno con hasta cuatro adatomos prefieren estructuras compactas en 2D con energías de unión relativamente bajas. El equipo determinó que el efecto de las regiones de interfaz y vértice en el comportamiento de migración de los grupos es significativamente fuerte en comparación con el tamaño de las nanopartículas.
Los mecanismos de migración son muy diferentes cuando los grupos están ubicados en el centro de la nanopartícula y cerca de las áreas de interfaz o vértice. Cerca de las interfaces y las áreas de los vértices, los átomos del sustrato tienden a participar en los procesos de migración de los cúmulos, y puede unirse a los adatomos para formar un grupo más grande o conducir a la disociación de un grupo a través del mecanismo de intercambio, lo que da como resultado que el adatom cruce las facetas.
Las barreras de energía calculadas para los trímeros sugieren que la migración concertada es más probable que el salto sucesivo de un solo adatom en los grupos.
El método computacional multiescala, desde el cálculo ab initio hasta el método de dinámica a largo plazo, se empleará además para estudiar la evolución estructural de grupos de metales de tamaño nanométrico con el aumento de tamaño y transformación de fase de estos grupos de metales. Estos estudios proporcionarán información importante sobre los catalizadores a nanoescala, sensores y aplicaciones electrocrómicas, como el vidrio inteligente, en las que las propiedades de transmisión de luz o calor del vidrio se modifican mediante la aplicación de voltaje.