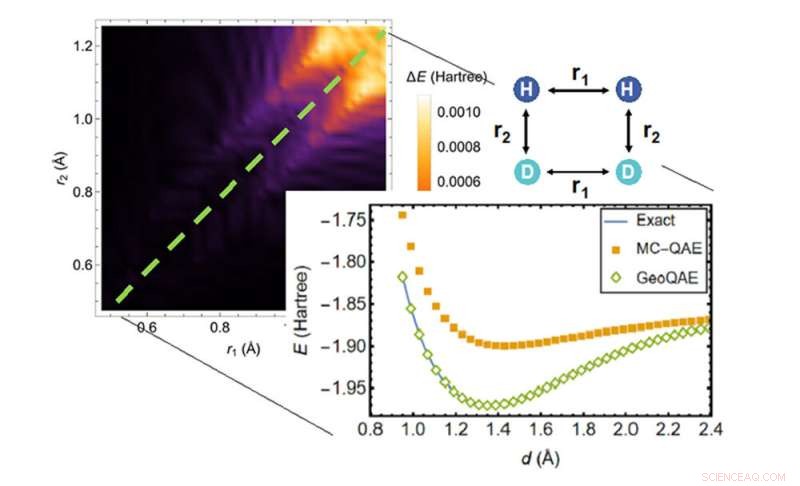
Al calcular la superficie de energía potencial de la reacción química de H2;+ D2 → 2HD, el nuevo algoritmo (rombos verdes) supera al algoritmo anterior (cuadrados naranjas) para encontrar la solución más precisa (línea azul). Crédito:Laboratorio Nacional de Brookhaven
Un equipo de investigadores del Laboratorio Nacional Brookhaven del Departamento de Energía de EE. UU. (DOE) y la Universidad Stony Brook han ideado un nuevo algoritmo cuántico para calcular las energías más bajas de las moléculas en configuraciones específicas durante las reacciones químicas, incluso cuando se rompen sus enlaces químicos. Como se describe en Investigación de revisión física , en comparación con algoritmos existentes similares, incluido el método anterior del equipo, el nuevo algoritmo mejorará significativamente la capacidad de los científicos para calcular con precisión y confiabilidad la superficie de energía potencial en las moléculas que reaccionan.
Para este trabajo, Deyu Lu, físico del Centro de Nanomateriales Funcionales (CFN) en Brookhaven Lab, trabajó con Tzu-Chieh Wei, profesor asociado especializado en ciencia de la información cuántica en el C.N. Instituto Yang de Física Teórica de la Universidad de Stony Brook, Qin Wu, teórico de CFN, y Hongye Yu, Ph.D. estudiante en Stony Brook.
"Comprender la mecánica cuántica de una molécula, cómo se comporta a nivel atómico, puede proporcionar información clave sobre sus propiedades químicas, como su estabilidad y reactividad", dijo Lu.
Una propiedad particular que ha sido un desafío para determinar es el estado fundamental de una molécula:el punto donde la energía electrónica total de la molécula (incluida la energía cinética y potencial) está en su punto más bajo y nada fuera de ese "sistema molecular" está excitando o cargando la molécula. electrones Cuando la estructura atómica de un sistema químico se vuelve más compleja, como en una molécula grande, pueden interactuar muchos más electrones. Esas interacciones hacen que calcular el estado fundamental de moléculas complejas sea extremadamente difícil.
El nuevo algoritmo cuántico mejora el algoritmo anterior para abordar este problema de una manera creativa. Aprovecha una deformación geométrica suave que se produce al variar continuamente las longitudes de los enlaces o los ángulos de los enlaces en la estructura de la molécula. Con este enfoque, los científicos dicen que pueden calcular el estado fundamental de las moléculas con mucha precisión, incluso cuando los enlaces químicos se rompen y se reforman durante las reacciones químicas.
Construyendo las bases
"Cuando se confía únicamente en los métodos informáticos tradicionales, este problema del estado fundamental contiene demasiadas variables para resolver, incluso en las supercomputadoras más poderosas", dijo Lu.
Puede pensar en un algoritmo como un conjunto de pasos para resolver un problema en particular. Las computadoras clásicas pueden ejecutar algoritmos complejos, pero a medida que se vuelven más grandes y más complicados, pueden volverse demasiado difíciles o lentos para que las computadoras clásicas los resuelvan de manera factible. Las computadoras cuánticas pueden acelerar el proceso aprovechando las reglas de la mecánica cuántica.
En la informática clásica, los datos se almacenan en bits que tienen un valor de 1 o 0. Un bit cuántico, conocido como qubit, puede tener un valor más allá de 0 o 1, incluso puede tener un valor de 0 y 1, en un la llamada superposición cuántica. En principio, estos qubits más "flexibles" pueden almacenar una mayor cantidad de información que los bits clásicos. Si los científicos pueden encontrar formas de aprovechar la capacidad de transporte de información de los qubits, el poder de cómputo puede expandirse exponencialmente con cada qubit adicional.
Los qubits, sin embargo, son bastante frágiles. A menudo pueden fallar cuando se extrae la información. Cuando un dispositivo cuántico interactúa con el entorno que lo rodea, puede generar ruido o interferencia que destruye el estado cuántico. Los cambios de temperatura, las vibraciones, las interferencias electromagnéticas e incluso los defectos materiales también pueden hacer que los qubits pierdan información.
Para compensar estos inconvenientes, los científicos desarrollaron una solución híbrida que aprovecha ambos algoritmos informáticos clásicos, que son más estables y prácticos.
Lu y Wei comenzaron a investigar sobre enfoques híbridos de computación cuántica y clásica en 2019. Esta subvención anual promueve la colaboración entre el Laboratorio Nacional de Brookhaven y la Universidad de Stony Brook al financiar iniciativas de investigación conjuntas que se alinean con las misiones de ambas instituciones. Con este trabajo inicial, Lu y Wei primero se enfocaron en resolver el problema del estado fundamental al reemplazar los algoritmos clásicos más "caros", los que eran mucho más complejos y requerían muchos más pasos (y más tiempo de computación) para completarse, con algoritmos cuánticos. .
Estirando lazos, creando nuevos caminos
Los investigadores señalan que todos los algoritmos cuánticos existentes tienen inconvenientes para resolver el problema del estado fundamental, incluido el que Wei y Yu desarrollaron en 2019. Si bien algunos algoritmos populares son precisos cuando una molécula está en su geometría de equilibrio:su disposición natural de átomos en tres dimensiones:esos algoritmos pueden volverse poco confiables cuando los enlaces químicos se rompen a grandes distancias atómicas. La formación y disociación de enlaces juegan un papel en muchas aplicaciones, como predecir cuánta energía se necesita para iniciar una reacción química, por lo que los científicos necesitaban una forma de abordar este problema a medida que reaccionan las moléculas. Necesitaban nuevos algoritmos cuánticos que pudieran describir la ruptura de enlaces.
Para esta nueva versión del algoritmo, el equipo trabajó con el Centro de codiseño para Quantum Advantage (C2QA) dirigido por Brookhaven-Lab, que se formó en 2020. Wei contribuye al impulso del software del centro, que se especializa en algoritmos cuánticos. El nuevo algoritmo del equipo utiliza un enfoque adiabático, que realiza cambios graduales, pero con algunas adaptaciones que aseguran que siga siendo confiable cuando se rompen los enlaces químicos.
"Un proceso adiabático funciona adaptando gradualmente las condiciones de un sistema mecánico cuántico", explicó Lu. "En cierto modo, está alcanzando una solución en pasos muy pequeños. El sistema evoluciona desde un modelo simple y solucionable hasta el objetivo final, generalmente un modelo más difícil. Sin embargo, además del estado fundamental, un sistema de muchos componentes electrónicos tiene muchos estados excitados a energías más altas. Esos estados excitados pueden representar un desafío cuando se utiliza este método para calcular el estado fundamental".
Wei comparó un algoritmo adiabático con conducir por una autopista, "si viajas de un pueblo a otro, hay varios caminos para llegar allí, pero quieres encontrar el más seguro y eficiente".
En el caso de la química cuántica, la clave es encontrar una "brecha de energía" lo suficientemente grande entre el estado fundamental y los estados excitados donde no existen estados electrónicos. Con un espacio lo suficientemente grande, los vehículos en la metáfora de la autopista no "cruzarán los carriles", por lo que sus caminos se pueden rastrear con precisión.
"Una brecha grande significa que puedes ir más rápido, así que, en cierto sentido, estás tratando de encontrar una carretera menos concurrida para conducir más rápido sin chocar contra nada", dijo Wei.
"Con estos algoritmos, la entrada de la ruta es una solución simple y bien definida de la computación clásica", señaló Wei. "También sabemos dónde está la salida, el estado fundamental de la molécula, y estábamos tratando de encontrar una forma de conectarla con la entrada de la manera más natural, una línea recta.
"Hicimos eso en nuestro primer artículo, pero la línea recta tenía obstáculos causados por el cierre de la brecha de energía y el cruce de caminos. Ahora tenemos una mejor solución".
Cuando los científicos probaron el algoritmo, demostraron que, incluso con cambios de longitud de enlace finitos, la versión mejorada seguía funcionando con precisión para el estado fundamental.
"Fuimos más allá de nuestra zona de confort, porque la química no es nuestro enfoque", dijo Wei. "Pero fue bueno encontrar una aplicación como esta y fomentar este tipo de colaboración con CFN. Es importante tener diferentes perspectivas en la investigación".
Señaló el esfuerzo acumulado de muchas personas. "En general, creo que estamos haciendo una pequeña contribución, pero esto podría ser la base para otro trabajo en estos campos", dijo. "Esta investigación no solo es fundamental, sino también una gran ilustración de cómo diferentes instituciones e instalaciones pueden unirse para aprovechar sus áreas de especialización". Hacia una computadora cuántica que calcule la energía molecular