Densidades electrónicas de estados (DOS) en varias etapas del proceso de compresión Crédito:@Michele Ceriotti
La combinación de cálculos de estructura electrónica y técnicas de aprendizaje automático (ML) se ha convertido en un enfoque común en el modelado atomístico de la materia. El uso de las dos técnicas juntas ha permitido a los investigadores, por ejemplo, crear modelos que utilicen coordenadas atómicas como las únicas entradas para predecir de forma económica cualquier propiedad que pueda calcularse mediante los cálculos de los primeros principios que se han utilizado para entrenarlos.
Si bien los primeros y hasta ahora más avanzados esfuerzos se han centrado en el uso de predicciones de energías totales y fuerzas atómicas para construir potenciales interatómicos, Los esfuerzos más recientes se han centrado en propiedades adicionales de los cristales y moléculas, como las energías de ionización, Blindajes químicos NMR, propiedades de respuesta dieléctrica y densidad de carga. En el artículo "Aprendiendo la densidad electrónica de estados en materia condensada, "Ceriotti y sus colegas se centran en la densidad electrónica de estados (DOS), otra cantidad que subyace a muchas propiedades útiles de los materiales, algunos de los cuales se pueden observar experimentalmente.
El DOS es esencialmente el número de estados diferentes que los electrones pueden ocupar a un nivel de energía particular, y se puede utilizar, por ejemplo, Calcular la contribución electrónica a la capacidad calorífica en metales y la densidad de portadores de carga libre en semiconductores. Es un proxy indirecto de propiedades como la banda prohibida de energía, la energía de la banda y el espectro de absorción óptica.
"Predecir el DOS es un ejercicio interesante en sí mismo porque es esencialmente la descripción más simple posible de la estructura electrónica más allá de la imagen del estado fundamental, ", Dijo Ceriotti." También es útil porque hay muchas propiedades que se pueden calcular a partir del DOS, lo que lo convierte en un gran ejemplo de cómo la próxima generación de modelos ML se puede utilizar de manera similar a los cálculos de estructura electrónica, usándolos de manera indirecta para calcular cantidades intermedias que luego pueden procesarse fácilmente para evaluar propiedades que son más difíciles de aprender directamente ".
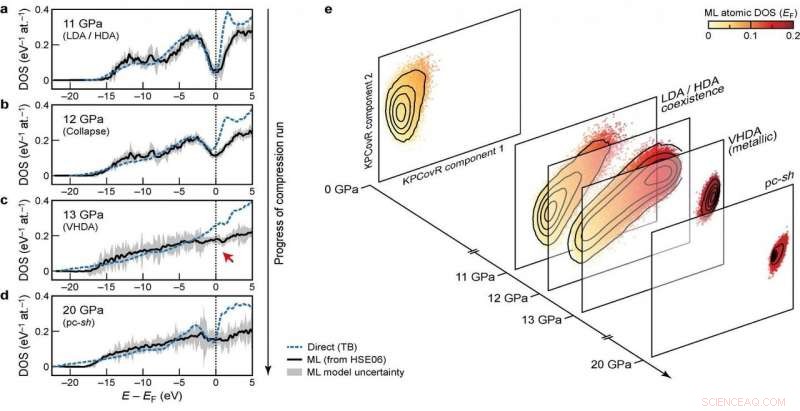
Al desarrollar el modelo, el grupo buscó asegurar la transferibilidad a través de diferentes fases, así como la escalabilidad a sistemas de gran tamaño. Su enfoque definitivo, que analiza cómo las diferentes configuraciones atómicas afectan la distribución de los niveles de energía, cumple estos objetivos:fue capaz de aprender y predecir DOS calculado con DFT para un conjunto de datos diverso de estructuras de silicio, cubriendo una amplia gama de condiciones termodinámicas y diferentes fases. También escala linealmente, en lugar de con el cubo del número de átomos como con los cálculos de estructura electrónica, haciéndolo aplicable a grandes estructuras. Finalmente, el modelo permitió un análisis del DOS local, dando a los investigadores la oportunidad de examinar la interacción entre los motivos estructurales y la estructura electrónica.
La combinación de transferibilidad, y escalabilidad de las predicciones a sistemas de gran tamaño, hacer que el modelo sea aplicable para abordar cuestiones abiertas de larga data en la ciencia de los materiales. El nuevo marco ya se ha utilizado para dilucidar las propiedades electrónicas de una simulación de 100.000 átomos de silicio amorfo, experimentando una serie de transiciones de fase cuando se comprime a 20 Gpa, en un artículo publicado en Naturaleza hoy en colaboración con un equipo formado por investigadores de Oxford, Cambridge, el Laboratorio de Investigación Naval de los Estados Unidos y la Universidad de Ohio. El DOS predicho también se utiliza para explicar cómo las transformaciones estructurales inducidas por la presión se acoplan a la estructura electrónica del material.
La combinación del nuevo modelo con uno de los modelos de energía potencial bien establecidos también permite calcular las contribuciones electrónicas a las propiedades macroscópicas, como la capacidad calorífica de los metales, y realizar simulaciones que tienen en cuenta los efectos de la temperatura electrónica finita, como se demostró. en otro artículo que se publicará próximamente sobre las propiedades del níquel a altas temperaturas. En efecto, El nuevo modelo es un paso crítico hacia el objetivo de MARVEL de desarrollar modelos integrados de aprendizaje automático que aumenten, y quizás eventualmente reemplacen, los costosos cálculos de estructuras electrónicas.
"Hay otras propiedades además de la densidad electrónica de los estados, como excitaciones ópticas, y respuesta de RMN, que hemos podido predecir con precisión con el aprendizaje automático ". Ceriotti dijo." Si podemos usarlos todos en combinación con potenciales interatómicos baratos y precisos, nos permitirá describir todas las propiedades de los materiales con la misma precisión lograda con cálculo de estructura, pero a una pequeña fracción del costo ".