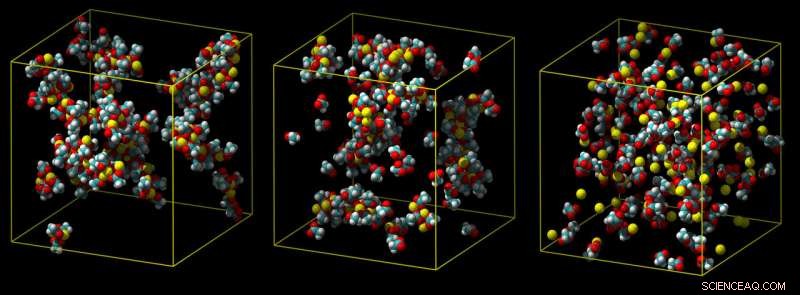
Un modelo de calcio estándar sobreestima la fuerza con la que se une el calcio, dando lugar a grupos de pares de iones (izquierda). Un modelo intermedio muestra menos agrupamiento (medio), y un modelo de escala de carga refinado predice correctamente una asociación débil con grupos carboxílicos en el agua (no se muestra) (derecha). Crédito:Philip Mason y Elise Duboue-Dijon
El calcio es esencial para el funcionamiento de nuestro cuerpo. Los iones de calcio permiten que las células se comuniquen entre sí, permitiendo que las neuronas interactúen, músculos para contraerse, y las células musculares del corazón para sincronizarse y latir. Para comprender mejor estos procesos, en el que los iones de calcio interactúan con moléculas biológicas como proteínas, los investigadores suelen utilizar simulaciones por ordenador. Pero los modelos precisos son desafiantes y computacionalmente costosos.
"Si tiene el modelo incorrecto de calcio, simplemente no funcionará, ", dijo Pavel Jungwirth del Instituto de Química Orgánica y Bioquímica de la Academia Checa de Ciencias en Praga." La mayoría de los modelos que están disponibles no son lo suficientemente precisos para capturar las características importantes del ion calcio ".
En el número de esta semana de La Revista de Física Química , sin embargo, El grupo de investigación de Jungwirth demuestra cómo una simple modificación en un modelo de computadora conduce a simulaciones altamente precisas, que sirven como herramientas poderosas para estudiar una variedad de procesos biológicos. "Creo que tenemos lo mejor de los modelos simples de calcio en el mundo en este momento, "Dijo Jungwirth.
Los iones de calcio viajan de una célula a otra como mensajeros. Cuando llegan a una celda, se unen a una molécula, como una proteína, desencadenando una cascada de respuestas químicas. Pero debido al ambiente acuoso del ion, simular exactamente cómo se une el calcio es difícil.
El ion calcio, que tiene doble carga positiva, interactúa fuertemente con los oxígenos de las moléculas de agua circundantes. Estos oxígenos tienen una carga negativa parcial (como en la molécula de agua) y el átomo de oxígeno atrae los electrones de los enlaces con mayor eficacia. Las fuerzas electrostáticas entre el calcio y el agua inducen a las moléculas de agua a reorganizarse alrededor del ion. El ion calcio también fuerza a los electrones en la molécula de agua a desplazarse, un fenómeno llamado polarización electrónica.
La mayoría de las simulaciones incorporan el reordenamiento de moléculas de agua. Pero debido a que calcular exactamente cómo se mueven los electrones requiere demasiada potencia de cálculo, no tienen en cuenta la polarización electrónica. Sin polarización electrónica, Jungwirth dijo:las simulaciones que involucran calcio son inexactas.
Típicamente, las interacciones con las moléculas de agua funcionan para alejar un ión de calcio de la molécula con la que está tratando de unirse, como en un tira y afloja molecular. Si una simulación no tiene completamente en cuenta estos efectos, sobreestima la fuerza con la que se une el calcio, produciendo iones que no pueden desprenderse, lo cual es poco realista.
Hace unos pocos años, sin embargo, Alexei Stuchebrukhov e Igor Leontyev propusieron una solución:bajar la carga eléctrica de los iones en las simulaciones. Resulta que escalar la carga en un factor de aproximadamente 0,75 imita el efecto de la polarización electrónica. Una escala tan simple tampoco agrega ninguna carga computacional adicional.
"Es casi un milagro, ", Dijo Jungwirth." Sabemos que no es una solución perfecta, pero tal vez resuelva el 90 por ciento del problema ".
Previamente, El equipo de Jungwirth probó la estrategia modelando la interacción relativamente simple entre los iones de calcio y cloruro. Para comprobar si las simulaciones eran precisas, y si la escala funcionaba, explotaron soluciones reales de cloruro de calcio con neutrones. Midiendo cómo esos neutrones se dispersaron del cloruro de calcio acuoso, los investigadores dedujeron su estructura y compararon los datos con las simulaciones.
En el nuevo estudio, los investigadores probaron su modelo con grupos carboxílicos, grupos moleculares que se encuentran en proteínas, y por tanto más relevante para la biología. Después de ajustar también la carga del grupo carboxílico, nuevamente demostraron que sus simulaciones coincidían muy bien con los datos de los experimentos de dispersión de neutrones.
Debido a que los grupos carboxílicos son simples en comparación con, decir, una proteína entera, los investigadores también pudieron describir las interacciones del calcio utilizando cálculos de estructura electrónica precisos pero computacionalmente costosos. Comparando estos cálculos con las simulaciones, nuevamente confirmaron la precisión de sus modelos.
Estas pruebas muestran que el nuevo modelo puede simular interacciones de calcio con casi cualquier proteína, Dijo Jungwirth. Los investigadores también han desarrollado un modelo análogo que funciona para las interacciones del calcio con los fosfolípidos en la membrana celular. El siguiente paso, él dijo, es hacer lo mismo con las moléculas de ADN y ARN. Y mas adelante los investigadores planean desarrollar un modelo similar para el magnesio, otro importante ion de señalización con sus propios desafíos únicos.