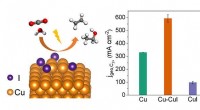
El equipo de investigación empleó cálculos de la teoría funcional de la densidad (DFT) para investigar el comportamiento de las moléculas de agua en presencia de iones. DFT es una poderosa herramienta para simular la estructura electrónica de materiales y moléculas. El equipo se centró en dos sistemas específicos:la solvatación de iones de litio (Li+) y la interacción de moléculas de agua con una superficie de litio-oxígeno (Li-O).
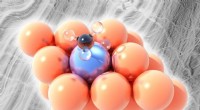
En el caso de la solvatación de Li+, los cálculos de DFT revelaron que las moléculas de agua forman una capa de hidratación altamente ordenada alrededor del ion litio. Esta estructura ordenada resulta de las fuertes interacciones electrostáticas entre el ion litio cargado positivamente y los átomos de oxígeno cargados negativamente de las moléculas de agua. La capa de hidratación protege eficazmente al ion de litio para que no interactúe con otros iones o moléculas en la solución, lo cual es crucial para estabilizar el ion y facilitar su transporte en reacciones electroquímicas.
Además, los investigadores estudiaron la interacción de las moléculas de agua con una superficie de Li-O, que representa la superficie del electrodo en una batería de iones de litio. Los cálculos de DFT mostraron que las moléculas de agua forman fuertes enlaces de hidrógeno con los átomos de oxígeno en la superficie, creando una red de agua que bloquea efectivamente la migración de iones de litio desde el electrodo. Este efecto de bloqueo es responsable de la pasivación de la superficie del electrodo y de la reducción del rendimiento de la batería con el tiempo.
En general, el marco teórico desarrollado por el equipo de investigación proporciona una comprensión integral del papel de las moléculas de agua en la síntesis de materiales y los procesos de fabricación que involucran iones. Los hallazgos contribuyen al diseño racional y la optimización de reacciones electroquímicas y sistemas de baterías, permitiendo materiales y dispositivos más eficientes y duraderos.