El equipo de Jianwei Shuai y el equipo de Jiahuai Han en la Universidad de Xiamen han desarrollado un software de análisis de datos de adquisición independiente de datos basado en codificador automático profundo para espectrometría de masas de proteínas, que realiza el análisis de péptidos y proteínas relevantes a partir de datos de espectrometría de masas de proteínas complejas y demuestra la superioridad y Versatilidad del método en diferentes instrumentos y muestras de especies. El estudio fue publicado en Research como "Estimado-DIA XMBD :codificador automático profundo para proteómica de adquisición independiente de datos".
Las proteínas desempeñan un papel fundamental como ejecutoras de las actividades de la vida celular, impulsando una gran variedad de procesos biológicos cruciales. En consecuencia, el campo de la proteómica ha recibido amplia atención. La proteómica implica el estudio integral de las propiedades de las proteínas, incluidas las modificaciones postraduccionales, los niveles de expresión de las proteínas, las interacciones proteína-proteína y más. Su objetivo general es obtener una comprensión holística de la patogénesis de las enfermedades, el metabolismo celular y otros procesos vitales a nivel de proteínas.
Entre las técnicas analíticas clave en la investigación proteómica, la espectrometría de masas de proteínas destaca como la más crítica. Con el tiempo, la tecnología de espectrometría de masas ha evolucionado para proporcionar a los investigadores herramientas confiables y dinámicas para el análisis proteómico.
Dos enfoques principales para la espectrometría de masas de proteínas son la adquisición dependiente de datos (DDA) y la adquisición independiente de datos (DIA). En DDA, todos los espectros de iones precursores de péptidos (MS1) se adquieren en modo de escaneo completo, seguido de la selección de los iones peptídicos con mayor contenido de N para la fragmentación y obtener espectros de iones de fragmentos (MS2).
A pesar de su utilidad, el DDA enfrenta desafíos relacionados con la reproducibilidad experimental y la detección de péptidos de baja abundancia debido a la aleatoriedad de la fragmentación de los péptidos y la selección preferencial de péptidos de alta intensidad.
Para superar estas limitaciones, se ha introducido el método de adquisición DIA. Esta técnica divide el rango de relación masa-carga de los espectros de iones originales en múltiples ventanas y fragmenta secuencialmente todos los péptidos dentro de cada ventana para obtener espectros de iones hijos. Un método DIA común es la adquisición de ventana secuencial de todos los iones de fragmentos teóricos (SWATH).
Si bien los datos de adquisición de DIA conservan información proteómica más completa, su gran tamaño de datos, su alta dimensionalidad y sus complejas señales espectrales plantean desafíos para su análisis. Como resultado, la minería de datos DIA se ha convertido en un foco importante en la comunidad proteómica.
El equipo de Jianwei Shuai y el equipo de Jiahuai Han colaboraron para desarrollar Dear-DIA, un software de análisis de datos de adquisición independiente de datos basado en aprendizaje profundo, que realiza la identificación de iones fragmentados correspondientes a diferentes péptidos de espectros de adquisición DIA complejos y demuestra la generalización a muestras complejas. de diferentes especies.
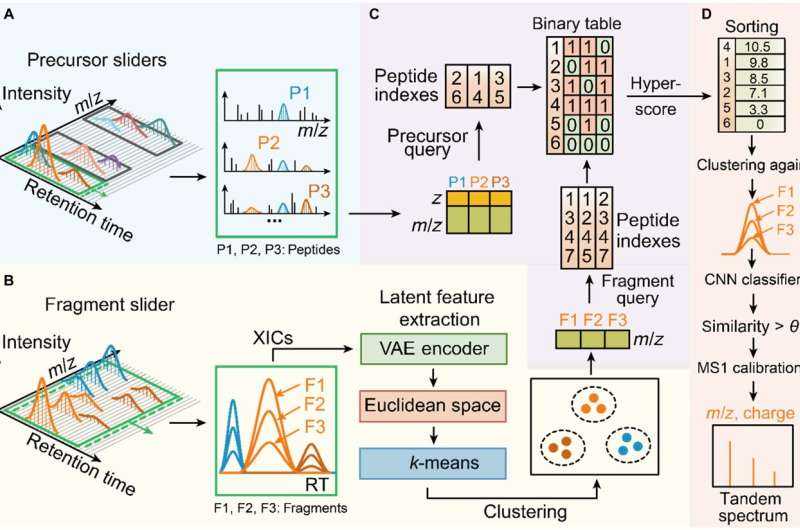
Dear-DIA primero divide los espectros en un control deslizante de ancho fijo con un ancho fijo a lo largo de la dirección del tiempo de retención (RT), y cada control deslizante contiene un conjunto de espectros precursores MS1 y espectros de fragmentos MS2 como unidad de procesamiento mínima. Luego, se utilizó un algoritmo de búsqueda de picos para eliminar los iones de fondo con baja relación señal-ruido y retener los iones precursores candidatos y los iones fragmentos candidatos.
A continuación, Dear-DIA utiliza un codificador automático variacional para extraer las características máximas de los iones de fragmentos y mapea las características en el espacio euclidiano, y luego agrupa las características, con diferentes clases de fragmentos correspondientes a diferentes péptidos, realizando así el proceso de deconvolución del espectrograma. P>
Dear-DIA incluye un algoritmo de indexación llamado PIndex, que relaciona los precursores con los resultados de agrupación de fragmentos y selecciona los mejores resultados de emparejamiento mediante puntuación. Dear-DIA utiliza una red neuronal convolucional para recalcular la similitud de la forma del pico de fragmentos de la misma clase para eliminar iones que interfieren y resultados de agrupación con baja similitud.
Los autores primero probaron el rendimiento de Dear-DIA en un conjunto de datos humanos de SGS que contenía 422 péptidos sintéticos de estándares marcados con isótopos estables divididos en 10 gradientes de dilución (de 1 a 512 veces de dilución), y los datos de DIA se obtuvieron en un AB. Espectrómetro de masas SCIEX TTOF5600 que utiliza la técnica SWATH para obtener datos de DIA.
Los resultados del análisis mostraron que Dear-DIA encontró más péptidos sintéticos en todas las soluciones diluidas en comparación con los dos métodos analíticos comúnmente utilizados, Spectronaut 14 y DIA-Umpire. Los autores también compararon la cantidad de péptidos y proteínas encontrados mediante los diferentes métodos analíticos para los conjuntos de datos SGS Human y L929 Mouse. Los resultados mostraron que Dear-DIA pudo encontrar más péptidos y proteínas en comparación con Spectronaut 14 y DIA-Umpire, cubriendo más del 85 % de sus resultados.
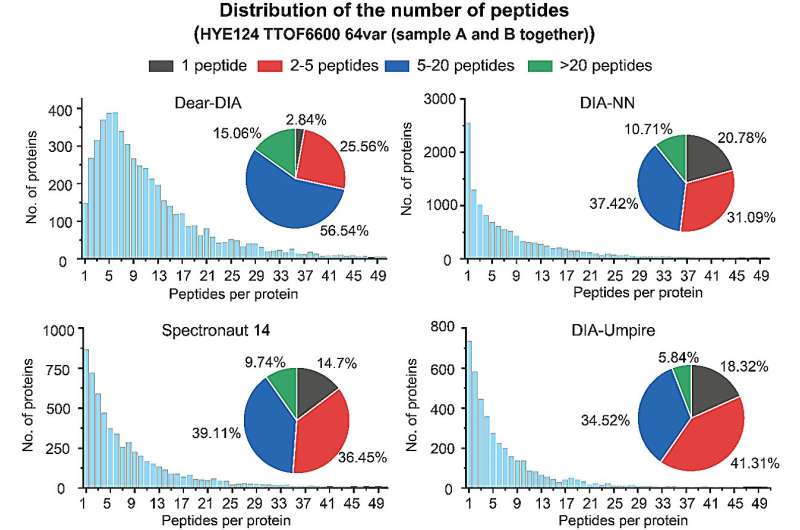
La confianza de los resultados del análisis proteómico también puede demostrarse mediante la cantidad de péptidos identificados para cada proteína. Las proteínas con 2 o más péptidos identificados generalmente se consideran identificaciones más creíbles. Los autores compararon la cantidad de proteínas versus péptidos reportados por Dear-DIA con el software existente en un conjunto de datos de especies mixtas (conjunto de datos HYE124 TTOF6600 64var).
El conjunto de datos contiene proteínas de tres especies:humana, de levadura y E. coli, y los datos se adquirieron en un espectrómetro de masas AB SCIEX TTOF6600 utilizando el método SWATH, con espectros de iones originales que contienen 64 ventanas variables. Los resultados del análisis mostraron que el 97,16% de las proteínas encontradas por Dear-DIA podrían corresponder a 2 o más péptidos, lo que es mucho más que DIA-NN, Spectronaut 14 y DIA-Umpire.
Las técnicas de adquisición de datos independientes para la proteómica se han adoptado ampliamente y los algoritmos de análisis relacionados se han convertido en un foco de investigación. El descubrimiento de proteínas a partir de datos masivos de espectrometría de masas es una tarea interesante y desafiante. En este artículo, el equipo desarrolló Dear-DIA, un software de análisis basado en aprendizaje profundo, que se utiliza para procesar una variedad de datos de adquisición de DIA altamente complejos y puede descubrir más péptidos y proteínas, además de reproducir la mayoría de los resultados de Espectronauta y árbitro DIA.
Además, aunque el conjunto de datos de entrenamiento proviene de E. coli, el excelente desempeño de Dear-DIA en el conjunto de datos de especies mixtas demuestra su fuerte capacidad de generalización para analizar datos proteómicos complejos. El aprendizaje profundo, como herramienta ampliamente utilizada para el análisis de big data, ha demostrado excelentes capacidades de minería de datos para descubrir asociaciones intrínsecas profundas en big data.
El uso del aprendizaje profundo para analizar datos de espectrometría de masas proteómica tiene un gran potencial y promoverá aún más el estudio de cuestiones fundamentales como las redes de señalización de proteínas.
Más información: Qingzu He y otros, Dear-DIA XMBD :El codificador automático profundo permite la deconvolución de la proteómica de adquisición independiente de datos, Investigación (2023). DOI:10.34133/investigación.0179
Información de la revista: Investigación
Proporcionado por Investigación