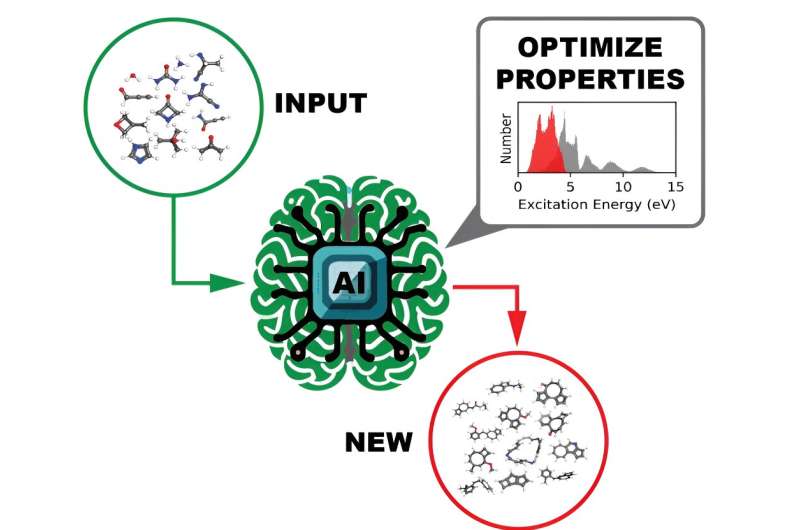
Un equipo de científicos computacionales del Laboratorio Nacional Oak Ridge del Departamento de Energía ha generado y publicado conjuntos de datos de una escala sin precedentes que proporcionan las propiedades espectrales ultravioleta visible de más de 10 millones de moléculas orgánicas. Comprender cómo interactúa una molécula con la luz es esencial para descubrir sus propiedades electrónicas y ópticas, que a su vez tienen posibles aplicaciones fotoactivas en productos como células solares o sistemas de imágenes médicas.
Utilizando recursos informáticos de alto rendimiento en Oak Ridge Leadership Computing Facility, el equipo de ORNL realizó cálculos de química cuántica para generar vastos conjuntos de datos. Para cada una de estas moléculas orgánicas, el equipo realizó cálculos de modelado de materiales atomísticos con varias aproximaciones para calcular diferentes propiedades de interés en estados excitados. Los hallazgos del equipo se publicaron en Scientific Data. .
El uso final previsto para los conjuntos de datos de código abierto es entrenar un modelo de aprendizaje profundo para identificar moléculas con propiedades optoelectrónicas y fotorreactividad personalizadas, un enfoque que es mucho más rápido y más fácil de realizar que los métodos actuales.
"El uso de modelos DL para el diseño molecular es esencial porque el espacio químico que debe explorarse para la búsqueda de estas moléculas es extremadamente grande", dijo el autor principal Massimiliano Lupo Pasini, científico de datos de la División de Ingeniería y Ciencias Computacionales del ORNL. P>
"Tanto los experimentos como los cálculos de primeros principios existentes, que se basan en las leyes físicas que determinan cómo interactúan la materia y la energía a nivel subatómico, son simplemente inasequibles por diferentes razones. Los experimentos requieren mucha mano de obra y los cálculos de primeros principios pueden fácilmente arruinar la supercomputación. instalaciones, pero los modelos DL proporcionan herramientas muy prometedoras para superar estas barreras", afirmó Lupo Pasini.
El proyecto despegó cuando Stephan Irle, líder del grupo de Química Computacional y Ciencias de Nanomateriales de ORNL, identificó los espectros ultravioleta-visible de las moléculas como una propiedad útil para predecir con modelos DL.
Construir un modelo DL lo suficientemente complejo como para identificar propiedades moleculares deseables requiere entrenarlo con enormes volúmenes de datos que exploren todas las diferentes regiones del espacio químico. Cuantos más datos se recopilen, más podrá el modelo DL entrenado en ellos lograr la solidez y generalización necesarias para funcionar de manera efectiva. Sin embargo, recopilar volúmenes tan grandes de datos científicos para DL escalable puede presentar problemas de flujo de datos, especialmente en instalaciones con múltiples usuarios como OLCF, una instalación para usuarios de la Oficina de Ciencias del DOE ubicada en ORNL.
"Un desafío que surge al generar grandes volúmenes de datos es que la cantidad de archivos a administrar aumenta drásticamente. Si no se administra correctamente, un volumen de datos tan grande puede comprometer el funcionamiento del sistema de archivos paralelo, que es un componente importante del estado. Instalaciones de HPC de última generación", afirmó Lupo Pasini.
Para abordar este desafío, Lupo Pasini colaboró con el científico informático de ORNL, Kshitij Mehta, para desarrollar un software de flujo de trabajo escalable que garantice que los archivos generados por el código de mecánica cuántica se manejen adecuadamente sin estresar el sistema de archivos, como Orion de OLCF, que es un sistema compartido. recurso que maneja la entrada, salida y almacenamiento de datos en sistemas de supercomputadoras.
Como prueba de concepto, el equipo generó el conjunto de datos GDB-9-Ex de 96.766 moléculas compuestas de carbono, nitrógeno, oxígeno y flúor, con como máximo nueve átomos distintos de hidrógeno. Demostró que el flujo de trabajo diseñado es eficaz y que el entrenamiento DL predice con precisión la posición y la intensidad de los picos más relevantes del espectro ultravioleta-visible.
A partir de ese éxito inicial, el equipo aumentó su volumen con el conjunto de datos ORNL_AISD-Ex, que contiene 10.502.917 moléculas compuestas de carbono, nitrógeno, oxígeno, flúor y azufre, con un máximo de 71 átomos distintos de hidrógeno. Pilsun Yoo, investigador postdoctoral asociado del grupo de Irle, desarrolló herramientas para analizar los conjuntos de datos resultantes.
El espectro ultravioleta-visible, que describe los modos de excitación de una molécula, se calculó para cada una de los más de 10 millones de moléculas. Esta información revela qué frecuencia de luz se requiere para apuntar a una molécula y romper algunos enlaces del compuesto químico.
Otra propiedad de interés calculada para cada molécula fue la brecha HOMO-LUMO (la brecha de energía entre el orbital molecular ocupado más alto y el orbital molecular desocupado más bajo) que mide de manera confiable la estabilidad de la molécula. Con esta información, un modelo DL podría examinar eficazmente los datos para identificar moléculas prometedoras para diferentes usos potenciales.
De hecho, Lupo Pasini y su equipo en ORNL, incluido el científico computacional en aprendizaje automático Pei Zhang y el científico investigador de datos HPC Jong Youl Choi, están desarrollando un modelo de DL:HydraGNN.
"La arquitectura HydraGNN toma la estructura atómica, la convierte en un gráfico y luego intenta predecir como resultado lo que produciría el código de primeros principios. Es un modelo sustituto para costosos cálculos de primeros principios", dijo Lupo Pasini.
Los resultados del entrenamiento de HydraGNN sobre los conjuntos de datos y sus descubrimientos moleculares se detallarán en un próximo artículo.
Más información: Massimiliano Lupo Pasini et al, Dos conjuntos de datos de estado excitado para espectros UV-vis químicos cuánticos de moléculas orgánicas, Datos científicos (2023). DOI:10.1038/s41597-023-02408-4
Información de la revista: Datos científicos
Proporcionado por el Laboratorio Nacional de Oak Ridge