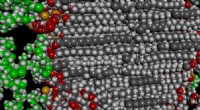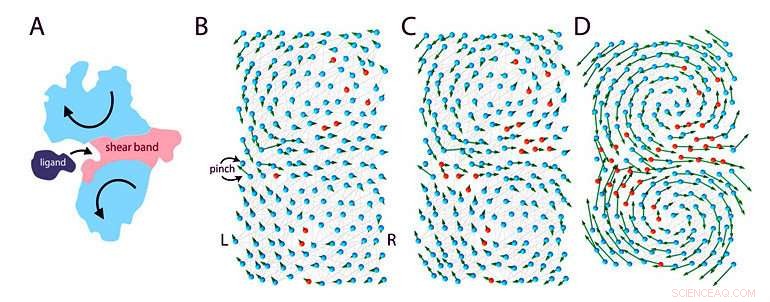
Figura 1:Modelo elástico de una proteína que se une a un ligando. (A) Cuando una proteína se une a un ligando, sufre un movimiento a gran escala (flechas) que son las firmas de las proteínas funcionales que se doblan. Esto es posible solo gracias a la presencia de ciertas regiones "flojas" ("banda de cizallamiento" rosa) a través de la proteína que separan las regiones rígidas (azules) de la proteína en dos dominios. (B) - (D) El equipo modeló una proteína de 200 aminoácidos durante diferentes etapas de evolución:pasando de un estado no funcional (B) a un estado funcional (D). La proteína se modela como una red de resorte elástica con dos tipos de aminoácidos, modelado como perlas:los aminoácidos rosas son flexibles y los aminoácidos azules son rígidos. Los investigadores imitan la evolución cambiando un aminoácido aleatorio en ese momento (mutación) de rosa a azul. Inicialmente, la proteína es en su mayoría rígida y no funcional. Durante la evolución, se añaden aminoácidos flexibles, algunos útiles, algunos no. Tiempo extraordinario, se forma una región "flácida" en el centro de la molécula, lo que hace que la proteína sea más flexible para doblarse y unirse al ligando. El modelo estimó que se alcanza una solución eficiente después de mil mutaciones. Crédito:Instituto de Ciencias Básicas
Un equipo internacional ha desarrollado un modelo que simula la evolución de las proteínas. Empezando desde rígido, proteínas no funcionales, el modelo informático muestra cómo los componentes de las proteínas en evolución pueden trabajar juntos para dar lugar a máquinas moleculares dinámicas y eficientes. La flexibilidad permite que las proteínas cambien su conformación 3D para unirse a otras moléculas:esta propiedad es crucial para su función. Prof. Tsvi Tlusty y Dr. Sandipan Dutta en el Center for Soft and Living Matter, dentro del Instituto de Ciencias Básicas (IBS, Corea del Sur), en colaboración con el Prof. Albert Libchaber de la Universidad Rockefeller y el Prof. Jean-Pierre Eckmann de la Universidad de Ginebra han imitado la evolución genética para obtener proteínas que pueden doblarse y unirse a otras moléculas. La comprensión de esta relación es uno de los aspectos más buscados de la biología de las proteínas; podría ayudar a explicar la acción farmacéutica de los fármacos que se unen a sus objetivos.
La evolución ha dado forma al mundo viviente que vemos a nuestro alrededor durante miles de millones de años. Millones de proteínas trabajan armoniosamente para mantener en marcha estos procesos vitales. Son responsables del buen funcionamiento de cualquier organismo:reconocen otras moléculas (ligandos), únete a ellos y conviértelos. Otros tienen función de transporte, proporcionar estructura, y apoyo a las células. Los genes almacenan la información sobre la producción y el diseño de estas máquinas moleculares. Sin embargo, a pesar de décadas de investigación, redactar el "mapa" que traza el camino desde los genes hasta la función de las proteínas no es trivial.
Según una hipótesis reciente, La función de las proteínas se basa en "articulaciones flexibles". Este estudio, publicado en procedimientos de la Academia Nacional de Ciencias ( PNAS ), examina el vínculo entre función y flexibilidad modelando proteínas como redes elásticas. En este modelo, las proteínas están formadas por aminoácidos flexibles (polares) y rígidos (hidrófobos) conectados por "resortes" moleculares. Si algunas regiones de la proteína son lo suficientemente flexibles, forman un canal "floppy", y toda la máquina molecular puede doblarse como una bisagra. Este movimiento les permite unirse eficazmente a otras moléculas. La unión entre un ligando y una proteína rígida o flexible se puede pensar como una pelota que aterriza sobre una roca o una almohada blanda. Es probable que la pelota rebote después de golpear la roca, pero es más probable que la almohada lo acepte. Por lo tanto, la proteína flexible es un mejor aglutinante.
En este modelo, los genes almacenan los detalles del diseño de la proteína de forma binaria:los aminoácidos flexibles se almacenan como ceros y los aminoácidos rígidos como unos. Como resultado, toda la estructura de la proteína se puede simplificar como un código, como 11110001 ... 111, similar a la memoria digital de una computadora. Sin embargo, no todos los códigos dan lugar a proteínas funcionales, por ejemplo, un código con solo unos:111111… 1111, daría lugar a una proteína completamente rígida, incapaz de moverse, y no funcional. Entre todos los códigos posibles, sólo algunos producen una proteína funcional con una región "flácida" en el centro que puede acoger al ligando.
El modelo imita la evolución cambiando un aminoácido aleatorio a la vez. Durante la evolución, los ceros y unos en el gen se invierten aleatoriamente a través de un proceso llamado mutación. La mayoría de las mutaciones no traen ninguna diferencia, o dar lugar a proteínas no funcionales, pero algunas mutaciones raras pueden dar lugar a una proteína más eficiente. Esencialmente, tanto las proteínas funcionales como las no funcionales se producen durante la evolución, pero según la teoría de Darwin de "supervivencia del más apto", sólo se conservan las proteínas funcionales y las proteínas no funcionales eventualmente se extinguen.
¿Qué aspecto tiene un código "funcional"? La respuesta no es sencilla. De hecho, el número de códigos de una proteína funcional, incluso una simple proteína, es enorme, más grande que el tamaño del universo. Sin embargo, utilizando técnicas de análisis de datos, es posible buscar patrones ocultos en todos los códigos funcionales para buscar algunas características unificadoras. Por ejemplo, el canal "flojo" en la proteína tiene características interesantes y peculiares, y una mutación en un extremo del canal tiene efectos de largo alcance que pueden afectar fuertemente el mantenimiento de mutaciones de otros aminoácidos distantes.
"En el futuro, planeamos explorar cómo aplicar este estudio a proteínas reales, como quinasas, "dijo el líder del grupo Tsvi Tlusty, un corresponsal en el estudio. "Es más, el estudio abre vías para investigar la evolución de otras funciones proteicas, como el reconocimiento molecular. Usando enormes bases de datos, que se han desarrollado a través de años de investigación, probablemente pueda descubrir algunos fenómenos subyacentes en la evolución de las proteínas ".