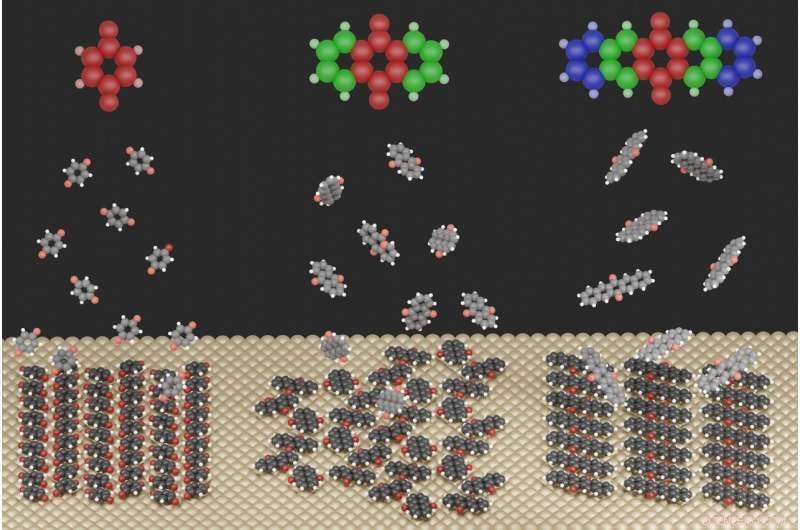
La ilustración muestra las estructuras de superficie fuertemente diferentes que se forman para las tres moléculas estudiadas cuando se adsorben en una superficie metálica. Crédito:Jeindl — TU Graz
Usando métodos de aprendizaje automático, Los investigadores de TU Graz pueden predecir la formación de estructuras de moléculas funcionalizadas en las interfaces de materiales híbridos. Ahora también han logrado mirar detrás de las fuerzas impulsoras de esta formación de estructura.
La producción de nanomateriales implica procesos de autoensamblaje de moléculas funcionalizadas (orgánicas) en superficies inorgánicas. Esta combinación de componentes orgánicos e inorgánicos es esencial para aplicaciones en electrónica orgánica y otras áreas de la nanotecnología.
Hasta ahora, A menudo se lograban determinadas propiedades superficiales deseadas mediante prueba y error. Las moléculas se modificaron químicamente hasta que se encontró el mejor resultado para la propiedad de superficie deseada. Sin embargo, los procesos que controlan el autoensamblaje de moléculas en las interfaces son tan complejos que pequeños cambios moleculares pueden conducir a motivos completamente diferentes. Los físicos de TU Graz explican esta inesperada formación de estructuras en un estudio publicado en la reconocida revista ACS Nano .
Para este propósito, los investigadores estudiaron compuestos de quinoides en una superficie plateada. Andreas Jeindl, primer autor del Instituto de Física del Estado Sólido, explica:"Ingenuamente, uno podría esperar que moléculas con tamaños ligeramente diferentes pero con la misma funcionalización formen motivos similares. En sorprendente contraste, Nuestro estudio teórico y experimental conjunto muestra que las quinonas pueden formar estructuras diversas. A pesar de las constantes condiciones iniciales, la formación de estas estructuras no se puede predecir ni planificar sin un conocimiento detallado de las interacciones relevantes ".
Tres fuerzas impulsoras opuestas
Los investigadores de Graz, junto con un equipo de la FSU Jena, ahora han comenzado a romper esta imprevisibilidad. Descubrieron que la formación de la estructura es el resultado de una compensación entre tres fuerzas impulsoras opuestas:la interacción entre las moléculas y el metal intenta forzar a todas las moléculas en la misma orientación, mientras que la interacción entre moléculas a veces favorece diferentes orientaciones. Las formas geométricas de las moléculas actúan entonces como un tercer factor, prevenir o permitir sólo parcialmente determinadas interacciones.
Basado en esto, pudieron establecer un principio de diseño con el cual las estructuras que se forman en las interfaces, y posteriormente sus propiedades, puede predecirse, al menos para una primera clase de moléculas. Un algoritmo de búsqueda (SAMPLE) basado en el aprendizaje automático juega un papel fundamental. Jeindl explica:"Pudimos mostrar en esta publicación que las estructuras predichas por nuestro algoritmo están en excelente acuerdo con las caracterizaciones experimentales de las interfaces orgánico-inorgánicas, tanto en cómo las moléculas se orientan en la superficie como en cómo se repiten los motivos en la superficie. superficie. Además, nuestro análisis, por primera vez, permitió un desglose detallado y cuantitativo de las fuerzas impulsoras, no solo de las estructuras formadas experimentalmente, pero de facto de todas las estructuras imaginables. Esta es una mirada importante entre bastidores de la formación de estructuras ".
Propiedades interfaciales con bloques de construcción modulares
La interacción no intuitiva de mecanismos de interacción igualmente importantes sigue siendo un desafío para el diseño de interfaces funcionales. Con una investigación detallada de todas las fuerzas impulsoras, sin embargo, No obstante, los físicos de TU Graz pueden idear un principio de diseño para el autoensamblaje de moléculas funcionalizadas para una clase determinada de moléculas. Una vez que haya suficientes análisis para diferentes clases de moléculas, las moléculas adecuadas para las propiedades interfaciales deseadas se pueden ensamblar fácilmente en la computadora a partir de bloques de construcción modulares.