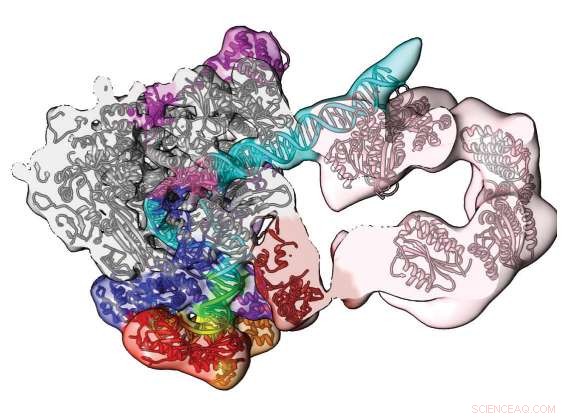
La supercomputación y la microscopía crioelectrónica revelan esta sección del complejo de preiniciación humana. El mapa y el modelo de densidad de conformación abiertos muestran la ruta del ADN (azul / verde) y su participación por el componente del factor de transcripción TFIIH (rosa). Reimpreso con permiso de Macmillan Publishers Ltd:He, Y. et al. Visualización de resolución casi atómica de la apertura del promotor de la transcripción humana. Naturaleza 533, 359–365 (2016).
Suena como algo salido de los Borg en Star Trek. Los robots de tamaño nanométrico se autoensamblan para formar máquinas biológicas que hacen el trabajo que lo mantiene vivo. Y, sin embargo, algo como esto realmente sucede.
Cada célula de nuestro cuerpo, ya sean de carne y hueso, cerebro y todo lo demás:tiene ADN idéntico, la escalera retorcida de ácidos nucleicos codificados de forma única para cada organismo. Conjuntos complejos que se asemejan a máquinas moleculares toman fragmentos de ADN llamados genes y producen una célula cerebral cuando es necesario. en lugar de, decir, una célula ósea. Estas máquinas moleculares son tan complejas, sin embargo, tan pequeño, que los científicos de hoy están empezando a comprender su estructura y función utilizando los últimos microscopios y supercomputadoras. Las máquinas moleculares biológicas podrían sentar las bases para desarrollar curas para enfermedades como el cáncer. Que pequeño se puede ver y que encontrara uno?
La microscopía crioelectrónica combinada con simulaciones de supercomputadora ha creado el mejor modelo hasta el momento, con detalles casi a nivel atómico, de una máquina molecular vital, el complejo de preiniciación humana (PIC). Un equipo científico de la Universidad Northwestern, Laboratorio Nacional de Berkeley, Universidad Estatal de Georgia, y UC Berkeley publicaron sus resultados en el PIC de mayo de 2016 en la revista Naturaleza .
"Por primera vez, Se han detallado las estructuras de los complejos grupos de moléculas que abren el ADN humano, "dijo el coautor del estudio Ivaylo Ivanov, profesor asociado de química en la Universidad Estatal de Georgia. Ivanov dirigió el trabajo computacional que modeló los átomos de las diferentes proteínas que actúan como engranajes de la máquina molecular PIC.
El PIC encuentra genes asociados con la producción de una proteína específica, como un anticuerpo o una enzima. Allí, el PIC separa las dos hebras de ADN y alimenta la hebra codificante a la enzima RNA polimerasa II del caballo de batalla. Esto inicia la transcripción, donde los bits de ADN son copiados por la ARN polimerasa II en una sola hebra de ARN mensajero. El ARN se abre camino hacia las 'fábricas de proteínas' en la célula llamadas ribosomas que los toman como órdenes para determinar qué proteína producir. Si el ADN es como el plano de una nueva casa, Los ARN son instrucciones para los 'contratistas' en la estación de trabajo de ribosomas. Las proteínas fabricadas son como las uñas, madera, yeso, y casi todo lo demás en la casa.
El experimento comenzó con imágenes cuidadosamente tomadas de PIC. Fueron realizados por un grupo liderado por la coautora del estudio Eva Nogales, profesor en el Departamento de Biología Molecular y Celular de UC Berkeley y también Científico Superior de la Facultad en el Laboratorio Nacional Lawrence Berkeley e Investigador Médico Howard Hughes.
El grupo de Nogales utilizó microscopía crioelectrónica (crio-EM), una estrella en ascenso en las técnicas de laboratorio. Congelaron criogénicamente el PIC humano unido al ADN. La congelación lo mantuvo en un estado químicamente activo, entorno casi natural. A continuación, lo atacaron con haces de electrones. Gracias a los avances recientes en la tecnología de detección directa de electrones, cryo-EM ahora puede obtener imágenes con una resolución casi atómica de estructuras biológicas grandes y complicadas que han demostrado ser demasiado difíciles de cristalizar. La técnica de referencia Cristalografía de rayos X, requiere muestras cristalizadas, y cryo-EM evita este difícil paso.
Se procesaron más de 1,4 millones de 'fotogramas congelados' crio-EM de PIC utilizando supercomputadoras en el Centro Nacional de Investigación Energética para Computación Científica para filtrar el ruido de fondo y mapas de densidad tridimensionales reconstruidos que muestran detalles en la forma de la molécula que nunca se habían hecho. visto antes.
"Cryo-EM está experimentando una gran expansión al igual que todo el software informático utilizado para generar los mapas de densidad y también para interpretarlos como lo hemos hecho en este estudio, "Nogales dijo." Nos permite obtener una mayor resolución de más estructuras en diferentes estados para que podamos describir no solo una imagen de cómo se ven, pero varias imágenes que muestran cómo se mueven. No vemos un continuo pero vemos instantáneas a través del proceso de acción ".
Los científicos del estudio luego construyeron un modelo preciso que tenía sentido físico de los mapas de densidad de PIC usando XSEDE, el entorno de descubrimiento de ciencia e ingeniería eXtream, financiado por la National Science Foundation. XSEDE permite a los científicos compartir de forma interactiva recursos informáticos, datos y experiencia a través de un único sistema virtual. El equipo de Ivaylo Ivanov ha realizado más de cuatro millones de horas centrales de simulaciones en la supercomputadora Stampede en el Centro de Computación Avanzada de Texas para modelar máquinas moleculares complejas. incluidos los de este estudio. El trabajo más amplio de máquinas moleculares de Ivanov también incluye una asignación XSEDE de 1,7 millones de horas centrales en la supercomputadora Comet en el Centro de Supercomputación de San Diego.
"He estado usando recursos XSEDE durante más de 12 años, ", Dijo Ivanov." Sin la disponibilidad de recursos XSEDE, toda nuestra investigación habría sido mucho más limitada en términos de los sistemas que podemos abordar. Para nosotros, XSEDE ha sido absolutamente esencial ".
El objetivo de todo este esfuerzo computacional es producir modelos atómicos que cuenten la historia completa de la estructura y función del complejo proteico de moléculas. Para llegar allí, el equipo de Ivanov tomó los doce componentes del ensamblaje PIC y creó modelos de homología para cada componente que explicaban sus secuencias de aminoácidos y su relación con estructuras tridimensionales de proteínas conocidas similares.
A continuación, aproximaron las densidades experimentales que el equipo de Nogales encontró en una cuadrícula. "Podemos utilizar un método llamado ajuste flexible de dinámica molecular, "explicó Ivanov, "donde esencialmente ejecutas una simulación de dinámica molecular. Y usas la densidad experimental para sesgar los átomos en la simulación de dinámica molecular para moverse a las regiones más densas del mapa EM. Ese es el proceso de ajuste flexible al mapa EM".
Refinaron el modelo con el paquete de refinamiento cristalográfico Phoenix. “Esa es una técnica complementaria que nos permite posicionar cadenas laterales y mejorar el modelo para que podamos capturar todos los detalles que están presentes en el mapa de densidad, "Dijo Ivanov.
XSEDE era "absolutamente necesario" para este modelado, dijo Ivanov. "Cuando incluimos agua y contraiones además del complejo PIC en una caja de simulación de dinámica molecular, obtenemos el tamaño del sistema de simulación de más de un millón de átomos. No se puede ejecutar eso en una estación de trabajo o incluso en un grupo modesto. Para eso realmente necesitamos ir a mil núcleos. En este caso, subimos a dos mil cuarenta y ocho núcleos. Y para eso necesitábamos acceso a Stampede, "Dijo Ivanov.
Una de las ideas obtenidas en el estudio es un modelo de trabajo de cómo PIC abre la doble hélice de ADN que de otro modo sería estable para la transcripción. Nogales explicó que uno podría imaginarse una cuerda hecha de dos hilos trenzados entre sí. Sostenga un extremo con mucha fuerza. Agarre el otro y gírelo en la dirección opuesta al enhebrado para desenredar el cordón. Básicamente, así es como lo hacen las máquinas vivientes que nos mantienen vivos.
"El ADN debe abrirse y trasladarse al sitio activo de la polimerasa para codificar el primer nucleótido de ARN, "explicó Nogales." El complejo de preiniciación mantiene las dos hebras del ADN muy juntas en un extremo, para que no puedan moverse y no puedan abrirse. Al otro lado del PIC hay una máquina que usa energía para impulsar el ADN, girándolo en la dirección opuesta en la que se enhebran los dos hilos. Y cuando esto suceda entre los dos lados, las hebras se abrirán, "dijo Nogales.
Este estudio resolvió la estructura de esa máquina molecular que actúa como los dedos que se retuercen, el componente del factor de transcripción TFIIH. "TFIIH tiene una subunidad translocase, cuya función es empujar simultáneamente el ADN hacia el sitio activo de la polimerasa y desenrollar el ADN. Por la combinación de empujar y desenrollar, efectivamente estás separando las dos hebras del ADN, "Dijo Ivanov.
Ambos científicos dijeron que apenas están comenzando a comprender la transcripción a nivel atómico, crucial para la expresión génica y, en última instancia, para la enfermedad. "Muchos estados patológicos surgen porque hay errores en la cantidad de lectura de un gen determinado y en la presencia de una proteína determinada con cierta actividad en la célula, "Nogales dijo." Esos estados de enfermedad podrían deberse a un exceso de producción de la proteína, o por el contrario no es suficiente. Es muy importante comprender el proceso molecular que regula esta producción para que podamos comprender el estado de la enfermedad ".
"Este trabajo ilustra bien dos principios generales que impulsarán la ciencia en los próximos años, "comentó Peter Preusch, oficial de programas de los Institutos Nacionales de Salud (NIH). "Uno es la aplicación de métodos híbridos:combinaciones de métodos biofísicos que incluyen cristalografía de rayos X y crioEM junto con métodos computacionales a gran escala para integrar información en complejos moleculares más grandes. Dos, Existe el requisito de que la ciencia en equipo aproveche la experiencia de varios investigadores para resolver problemas que no pueden ser abordados por un solo laboratorio trabajando solo ". Peter Preusch es el jefe de la rama de biofísica, División de Biología Celular y Biofísica, Instituto Nacional de Ciencias Médicas Generales, NIH.
Si bien este trabajo fundamental no produce curas directamente, sienta las bases para ayudar a desarrollarlos en el futuro, dijo Ivanov. "Para comprender la enfermedad, tenemos que entender cómo funcionan estos complejos en primer lugar… Una colaboración entre modeladores computacionales y biólogos estructurales experimentales podría ser muy fructífera en el futuro. "
El estudio Nature Articles de mayo de 2016 (DOI:10.1038 / nature17970), "Visualización de resolución casi atómica de la apertura del promotor de la transcripción humana, "fue escrito por Yuan He, Lawrence Berkeley National Laboratory y ahora en Northwestern University; Chunli Yan e Ivaylo Ivanov, Universidad Estatal de Georgia; Jie Fang, Carla Inouye, Robert Tjian, Eva Nogales, UC Berkeley. La financiación provino del Instituto Nacional de Ciencias Médicas Generales (NIH) y la Fundación Nacional de Ciencias.