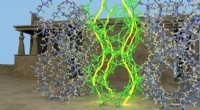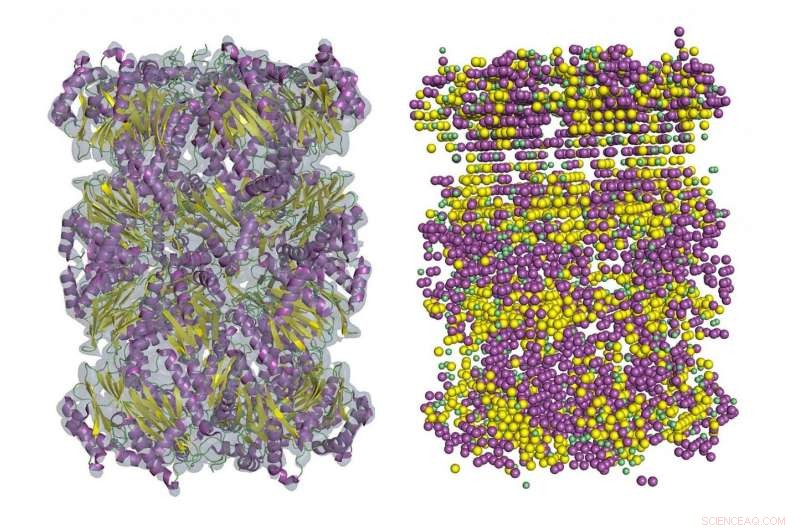
Un ejemplo de detección de estructura secundaria en mapa de densidad crio-EM usando Emap2Sec. A la izquierda se muestra un mapa EM del proteasoma archaeal 20S (EMDB ID:EMD-1733). A la derecha se detectan estructuras secundarias por Emap2Sec. Los puntos en magenta son las posiciones de las hélices alfa detectadas; los puntos amarillos son hebras beta detectadas, y los puntos verdes son para bobinas detectadas (otras estructuras). Crédito:imagen de la Universidad de Purdue / Daisuke Kihara
La microscopía crioelectrónica es ahora el método más popular para determinar las estructuras de proteínas, que ayuda a los investigadores a desarrollar medicamentos para diferentes tipos de dolencias. Durante las últimas décadas, ha reemplazado a la cristalografía de rayos X porque puede obtener imágenes de proteínas que no se pueden formar fácilmente en cristales grandes. La nueva técnica fue tan revolucionaria que le valió a sus desarrolladores el Premio Nobel de Química de 2017.
El producto final de cryo-EM es un mapa de la densidad de átomos en moléculas biológicas, pero para alcanzar el nivel de detalle que necesitan los investigadores, necesitan realizar un análisis más detallado. Un nuevo estudio en la revista Métodos de la naturaleza describe una técnica para llevar mapas de baja resolución a la par.
El enfoque que utilizan los investigadores para hacer esto depende del nivel de detalle con el que comienzan. Mapas de 2 a 3 ångström (Å, una unidad de longitud utilizada para expresar el tamaño de los átomos y las moléculas) generalmente se consideran de alta resolución. Sin embargo, mapas de esta calidad son difíciles de conseguir, y muchos todavía se producen comúnmente en el rango de 4 a 10 Å. De todas las proteínas depositadas en el banco de datos de microscopía electrónica de 2016-18, más del 50% se resolvieron con resolución intermedia.
"Si la resolución es mejor que tres, luego, las herramientas convencionales pueden rastrear la posición de los aminoácidos y construir un mapa de las posiciones de los átomos. Pero, con frecuencia, cryo-EM no puede proporcionarle un mapa de 3 Å, "dijo Daisuke Kihara, profesor de ciencias biológicas e informática en la Universidad de Purdue. "En mapas de 5 Å o menos, por lo general, no puede ver la conectividad en cadena en absoluto ".
Las proteínas son en realidad cadenas de aminoácidos, y la unión entre grupos amino y grupos carboxilo crea a veces ciertos patrones de plegamiento. Estos patrones conocidas como hélices alfa y hebras beta, forman la estructura secundaria de la proteína.
En mapas de 5 a 8 Å, algunos fragmentos de la estructura secundaria de las proteínas suelen ser visibles, pero rastrear toda la cadena sería muy difícil. El nuevo método de Kihara, conocido como Emap2sec, descubre estructuras secundarias en mapas de 6 a 10 Å.
Emap2sec tiene una red neuronal convolucional profunda en el núcleo de su algoritmo. Estas redes son sistemas de aprendizaje profundo que se utilizan principalmente para clasificar imágenes, agruparlos por similitud y realizar el reconocimiento de objetos. Funciona para la identificación de estructuras de proteínas en mapas 3-D porque el método "convoluciona" las características de densidad del mapa local en imágenes de una región más grande a medida que la información pasa a través de capas de la red neuronal. La predicción local se realiza en el contexto de una gran región del mapa.
Las estructuras secundarias identificadas en mapas 3D ayudan a los investigadores a asignar estructuras conocidas de proteínas que ya se han resuelto en el mapa. Esto significa que a veces tienen un punto de partida, o al menos una pista de cómo se ve parte de la estructura. Emap2sec puede ayudar a los investigadores a encajar su pieza en el rompecabezas de forma más rápida y sencilla. La información de la estructura identificada también puede ser útil para encontrar errores en el modelado de estructuras.