Un equipo dirigido por el profesor Frank Glorius del Instituto de Química Orgánica de la Universidad de Münster ha desarrollado un algoritmo evolutivo que identifica las estructuras de una molécula que son particularmente relevantes para una cuestión respectiva y las utiliza para codificar las propiedades de las moléculas para varios modelos de aprendizaje automático.
El método también es adecuado para la predicción mecánica de propiedades químicas cuánticas y la toxicidad de moléculas. Se puede aplicar a cualquier conjunto de datos moleculares y no requiere conocimientos expertos de las relaciones subyacentes.
La inteligencia artificial y el aprendizaje automático son cada vez más relevantes en la vida cotidiana, y lo mismo ocurre con la química. Los químicos orgánicos, por ejemplo, están interesados en cómo el aprendizaje automático puede ayudar a descubrir y sintetizar nuevas moléculas que sean efectivas contra enfermedades o útiles de otras maneras.
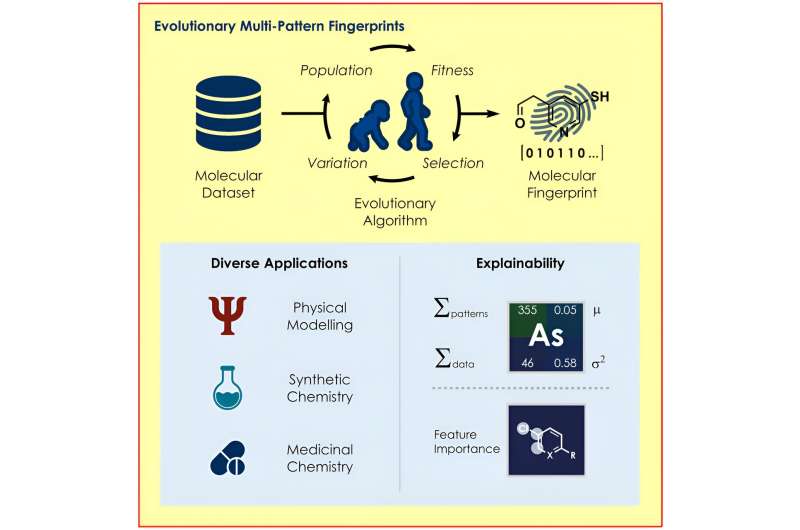
El nuevo algoritmo desarrollado por el equipo de Glorius busca representaciones moleculares óptimas basadas en los principios de la evolución, utilizando mecanismos como la reproducción, la mutación y la selección. Dependiendo del modelo y de la pregunta planteada, se crean "huellas dactilares moleculares" personalizadas, que los químicos utilizaron en su estudio para predecir reacciones químicas con una precisión sorprendente.
El método, publicado en la revista Chem , también es adecuado para predecir propiedades químicas cuánticas y la toxicidad de moléculas.
Para utilizar el aprendizaje automático, los investigadores primero deben convertir las moléculas a un formato legible por computadora. Muchos grupos de investigación ya han abordado este problema y, en consecuencia, existen diversas formas de realizar esta tarea. Sin embargo, es difícil predecir cuál de los métodos disponibles es mejor para responder una pregunta específica; por ejemplo, para determinar si un compuesto químico es dañino para los humanos.
El nuevo algoritmo está diseñado para ayudar a encontrar la huella molecular óptima en cada caso. Para ello, el algoritmo selecciona gradualmente las huellas moleculares que logran mejores resultados en la predicción a partir de muchas huellas moleculares generadas aleatoriamente.
"Siguiendo el ejemplo de la naturaleza, utilizamos mutaciones, es decir, cambios aleatorios en componentes individuales de las huellas dactilares, o recombinamos componentes de dos huellas dactilares", explica el estudiante de doctorado Felix Katzenburg.
"En otros estudios, las moléculas a menudo se describen mediante propiedades cuantificables que han sido seleccionadas y calculadas por humanos", añade Glorius.
"Dado que el algoritmo que desarrollamos identifica automáticamente las estructuras moleculares relevantes, no existen sesgos sistemáticos causados por expertos humanos".
Otra ventaja es que el método de codificación permite comprender por qué un modelo realiza una determinada predicción. Por ejemplo, es posible sacar conclusiones sobre qué partes de una molécula impactan positiva o negativamente en la predicción de cómo se desarrollaría una reacción, lo que permite a los investigadores cambiar las estructuras relevantes de manera específica.
El equipo de Münster descubrió que su nuevo método no siempre lograba los resultados más óptimos.
"Cuando se ha invertido una gran experiencia humana en la selección de propiedades moleculares particularmente relevantes o se dispone de grandes cantidades de datos, otros métodos, como las redes neuronales, a veces tienen la ventaja", afirma Katzenburg.
Sin embargo, uno de los objetivos principales del estudio fue desarrollar un método para codificar moléculas que pueda aplicarse a cualquier conjunto de datos moleculares y no requiera conocimientos expertos de las relaciones subyacentes.
Más información: Philipp M. Pflüger et al, Un algoritmo evolutivo para representaciones moleculares interpretables, Chem (2024). DOI:10.1016/j.chempr.2024.02.004
Información de la revista: Química
Proporcionado por la Universidad de Münster