Miembros de CAMERA (desde la izquierda) Peter Zwart, Jeff Donatelli y Kanupriya Pande, coautores de un artículo que describe cómo el algoritmo M-TIP del grupo determinaba las estructuras de virus 3D a partir de datos de difracción de una sola partícula. Donatelli sostiene un modelo impreso en 3D de uno de los virus reconstruidos por M-TIP. Crédito:Marilyn Chung, Laboratorio de Berkeley
Como parte de un equipo de investigación internacional, Jeff Donatelli, Peter Zwart y Kanupriya Pande del Centro de Matemáticas Avanzadas para Aplicaciones de Investigación Energética (CAMERA) en el Laboratorio Nacional Lawrence Berkeley (Berkeley Lab) contribuyeron con algoritmos clave que ayudaron a lograr un objetivo propuesto por primera vez hace más de 40 años:usar correlaciones angulares de rayos X instantáneas de moléculas no cristalinas para determinar la estructura 3D de objetos biológicos importantes. Esta técnica tiene el potencial de permitir a los científicos arrojar luz sobre la estructura biológica y la dinámica que antes era imposible de observar con los métodos tradicionales de rayos X.
El avance fue el resultado de un experimento de difracción de una sola partícula realizado en la fuente de luz coherente Linac (LCLS) del Departamento de Energía (DOE) por la Iniciativa de Partícula Única organizada por el Laboratorio Nacional de Aceleradores de SLAC. Como parte de esta iniciativa, el equipo de CAMERA combinó esfuerzos con Ruslan Kurta, un físico en la instalación europea XFEL (láser de electrones libres de rayos X) en Alemania, para analizar las correlaciones angulares de los datos experimentales y utilizar el algoritmo de fase iterativa de múltiples niveles (M-TIP) de CAMERA para realizar las primeras reconstrucciones de virus 3D exitosas a partir de correlaciones experimentales. Los resultados se describen en un artículo publicado el 12 de octubre en Cartas de revisión física .
"Durante los últimos 40 años, esto se consideró un problema que no se pudo resolver, "dijo Peter Zwart, coautor del artículo y biocientífico físico que es miembro de CAMERA de la División de Biofísica Molecular e Imágenes Integradas en Berkeley Lab. "Pero resulta que las herramientas matemáticas que desarrollamos son capaces de aprovechar la información adicional oculta en el problema que se había pasado por alto anteriormente. Es gratificante ver que nuestro enfoque teórico conduce a una herramienta práctica".
Nuevas oportunidades de investigación habilitadas por XFEL
Durante gran parte del siglo pasado, La técnica de referencia para determinar la estructura molecular de alta resolución ha sido la cristalografía de rayos X, donde la muestra de interés se organiza en una gran red periódica y se expone a rayos X que se dispersan y forman patrones de difracción que se recogen en un detector. Aunque la cristalografía ha tenido éxito en la determinación de muchas estructuras de alta resolución, Es un desafío utilizar esta técnica para estudiar estructuras que no son susceptibles a la cristalización o cambios estructurales que no ocurren naturalmente dentro de un cristal.
La creación de las instalaciones de XFEL, incluyendo la fuente de luz coherente Linac (LCLS) y el X-FEL europeo, han creado oportunidades para realizar nuevos experimentos que pueden superar las limitaciones de la cristalografía tradicional. En particular, Los rayos XFEL son varios órdenes de magnitud más brillantes que las fuentes de luz de rayos X tradicionales y tienen longitudes de pulso mucho más cortas. que les permiten recolectar señales de difracción medibles de muestras no cristalizadas más pequeñas y también estudiar la dinámica rápida. La difracción de una sola partícula es una de esas técnicas experimentales emergentes habilitadas por XFELS, donde se recolectan imágenes de difracción de moléculas individuales en lugar de cristales. Estas técnicas de una sola partícula se pueden utilizar para estudiar la estructura molecular y la dinámica que ha sido difícil de estudiar con las técnicas de imagen tradicionales.
Superar las limitaciones en la obtención de imágenes de una sola partícula mediante correlaciones angulares
Un desafío importante de la formación de imágenes de una sola partícula es el de la determinación de la orientación. "En un experimento de una sola partícula, no tiene control sobre la rotación de las partículas cuando son golpeadas por el haz de rayos X, por lo que cada instantánea de un éxito exitoso contendrá información sobre la muestra desde una orientación diferente, "dijo el coautor Jeff Donatelli, un matemático aplicado en CAMERA que desarrolló muchos de los algoritmos en el nuevo marco. "La mayoría de los enfoques para el análisis de partículas individuales se han basado hasta ahora en tratar de determinar estas orientaciones de partículas a partir de las imágenes; sin embargo, la mejor resolución que se puede lograr a partir de estos análisis está restringida por la precisión con la que se pueden determinar estas orientaciones a partir de datos ruidosos ".
En lugar de intentar determinar directamente estas orientaciones, el equipo adoptó un enfoque diferente basado en la idea propuesta originalmente en la década de 1970 por Zvi Kam. "En lugar de examinar las intensidades de datos individuales en un intento de encontrar la orientación correcta para cada cuadro medido, Eliminamos este paso por completo mediante el uso de las llamadas funciones de correlación cruzada, "Dijo Kurta.
Este enfoque, conocido como dispersión de rayos X por fluctuación, se basa en analizar las correlaciones angulares de ultracorto, intensos pulsos de rayos X dispersos de biomoléculas no cristalinas. "La belleza de usar datos de correlación es que contiene una huella digital completa de la estructura 3D de un objeto que amplía los enfoques tradicionales de dispersión de soluciones, "Dijo Zwart.
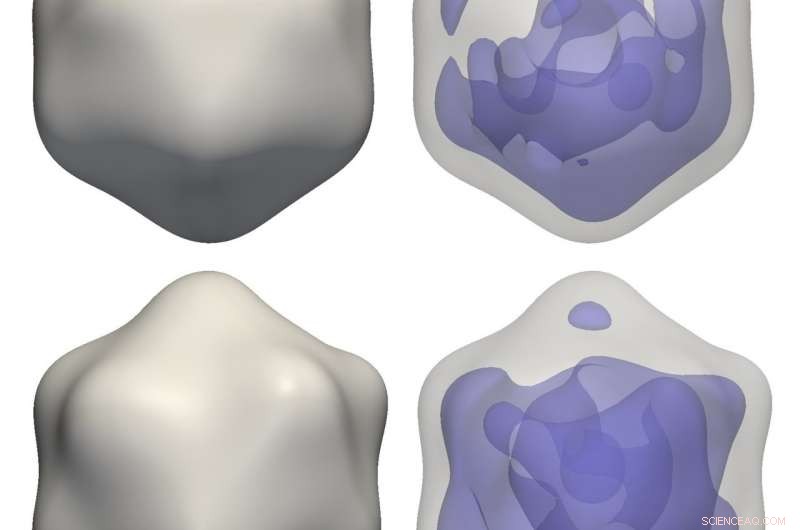
Virus reconstruidos:reconstrucciones de un virus enano del arroz (arriba) y un bacteriófago PR772 (abajo) a partir de datos de correlación experimental utilizando M-TIP. Las imágenes de la derecha muestran asimetrías en el material genético interno para cada reconstrucción de virus. Crédito:Jeff Donatelli, Laboratorio de Berkeley
Reconstrucción de la estructura 3D a partir de correlaciones con el algoritmo M-TIP de CAMERA
El avance del equipo en la reconstrucción de la estructura 3D a partir de datos de correlación fue posible gracias al algoritmo de fase iterativa de múltiples niveles (M-TIP) desarrollado por CAMERA. "Entre las ventajas destacadas de M-TIP está su capacidad para resolver la estructura directamente a partir de los datos de correlación sin tener que depender de ninguna restricción de simetría, y, más importante, sin necesidad de resolver el problema de determinación de la orientación, "Dijo Donatelli.
Donatelli, El director de CAMERA, James Sethian y Zwart, desarrollaron su marco M-TIP mediante el desarrollo de una generalización matemática de una clase de algoritmos conocidos como técnicas de fases iterativas, que se utilizan para determinar la estructura en un problema más simple, conocido como recuperación de fase. En agosto de 2015 se publicó un artículo que describe el marco M-TIP original en procedimientos de la Academia Nacional de Ciencias .
"Los análisis de correlación avanzados en combinación con reconstrucciones ab-initio por M-TIP definen claramente una ruta eficiente para el análisis estructural de objetos a nanoescala en XFELs, "Dijo Zwart.
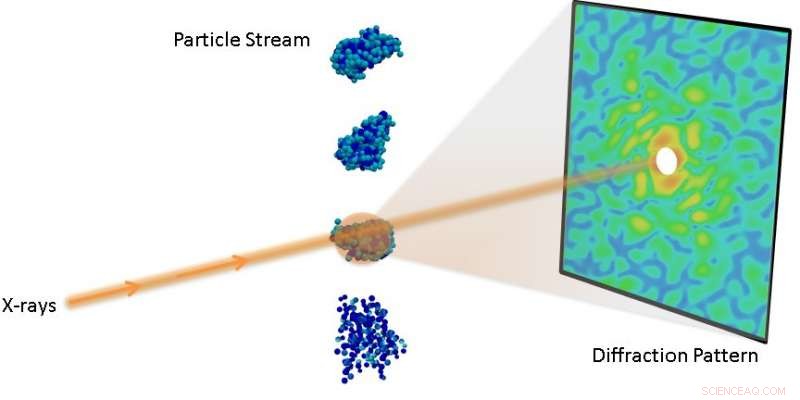
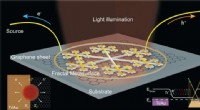
Configuración experimental para un experimento de difracción de una sola partícula. Crédito:Laboratorio Nacional Lawrence Berkeley
Direcciones futuras para el análisis de correlación y M-TIP
El equipo señala que los métodos utilizados en este análisis también se pueden aplicar para analizar los datos de difracción cuando hay más de una partícula por disparo.
"La mayoría de los algoritmos para la obtención de imágenes de una sola partícula solo pueden manejar una molécula a la vez, limitando así la señal y la resolución. Nuestro enfoque por otra parte, es escalable, por lo que también deberíamos poder medir más de una partícula a la vez, ", dijo Kurta. La obtención de imágenes con más de una partícula por disparo permitirá a los científicos lograr tasas de acierto mucho más altas, ya que es más fácil usar un haz ancho y golpear muchas partículas a la vez, y también evitará la necesidad de separar los impactos de una sola partícula de los impactos de múltiples partículas y los disparos en blanco, que es otro requisito desafiante en las imágenes tradicionales de una sola partícula.
Como parte del conjunto de herramientas computacionales de CAMERA, También han desarrollado una versión diferente de M-TIP que resuelve el problema de orientación y puede clasificar las imágenes en estados conformacionales, y, en consecuencia, se puede utilizar para estudiar pequeñas diferencias biológicas en la muestra medida. Esta versión alternativa de M-TIP se describió en un artículo publicado el 26 de junio de 2017 en la procedimientos de la Academia Nacional de Ciencias y es parte de una nueva iniciativa de colaboración entre SLAC National Accelerator Laboratory, CÁMARA, the National Energy Research Scientific Computing Center (NERSC) and Los Alamos National Laboratory as part of DOE's Exascale Computing Project (ECP).