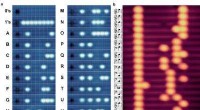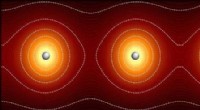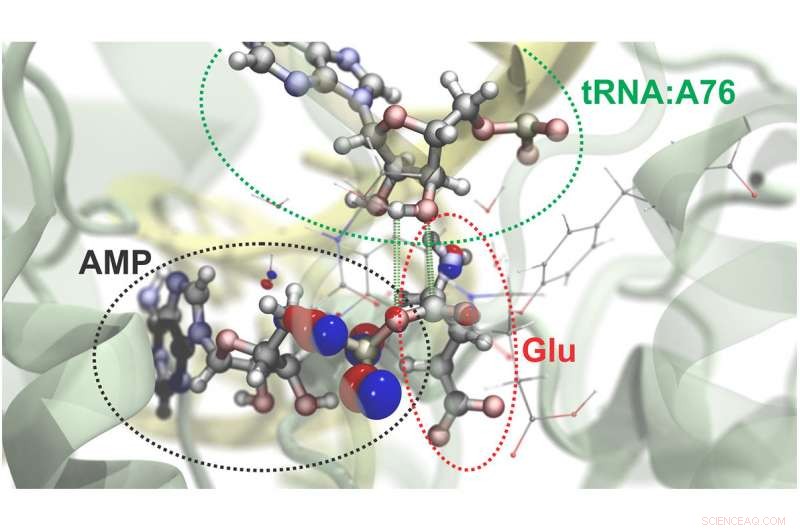
Los investigadores pueden simular dinámicas atómicas y subatómicas en grandes sistemas moleculares. A continuación se muestra una visualización del proceso mediante el cual el aminoácido glutamato (Glu) se une a una región específica de su ARN de transferencia (ARNt). Una molécula rica en energía, ATP, impulsa esta reacción y se convierte en AMP en el proceso. Las burbujas rojas y azules representan la probabilidad de encontrar electrones en regiones particulares. Las líneas de puntos verdes delinean los átomos que se unen en esta reacción química. Crédito:Rafael Bernardi, Zan Luthey-Schulten y Marcelo Melo
Los científicos han construido un "microscopio computacional" que puede simular las fuerzas atómicas y subatómicas que impulsan las interacciones moleculares. Esta herramienta agilizará los esfuerzos para comprender la química de la vida, Modelar grandes sistemas moleculares y desarrollar nuevos agentes farmacéuticos e industriales. dicen los investigadores.
Informan sus hallazgos en la revista. Métodos de la naturaleza .
Los científicos combinaron dos enfoques computacionales utilizados para simular interacciones moleculares. El primero, un programa de dinámica molecular a nanoescala conocido como NAMD, utiliza métodos de mecánica clásica para modelar la estructura y simular el comportamiento de cientos de millones de átomos individuales. El segundo programa se acerca al reino subatómico, simulando las interacciones de los protones, neutrones y electrones. El modelado a esta escala de la mecánica cuántica exige mucha potencia computacional, por lo que los investigadores implementaron un método para dividir moléculas grandes en regiones de mecánica cuántica y clásica. Esto les permite enfocar sus recursos computacionales en pequeñas regiones involucradas en interacciones críticas, como la formación o ruptura de enlaces químicos.
Tanto los programas de mecánica molecular como los de mecánica cuántica han estado disponibles durante años, y otros equipos han trabajado para combinarlos, dijo la profesora de química de la Universidad de Illinois Zaida (Zan) Luthey-Schulten, quien dirigió la nueva investigación con su esposo, U. de I. profesor de física Klaus Schulten. Pero el nuevo esfuerzo agiliza el proceso de creación, realizar y analizar las simulaciones.
"Lo configuramos para que los investigadores puedan elegir fácilmente cómo dividirán sus propios sistemas, ", Dijo Luthey-Schulten." Mis propios alumnos lo están probando, y la mayoría de ellos pueden hacerlo sin mucha dificultad ".
Schulten desarrolló NAMD en Illinois en 1995, combinándolo con un software de visualización, VMD, lo que permite a los investigadores observar el desarrollo de interacciones moleculares a gran escala. Schulten, que murió en 2016, equiparó este enfoque a "construir un microscopio computacional".
El microscopio computacional es ideal para modelar rasgos estructurales y movimientos de grandes complejos. Por ejemplo, en 2013, Schulten y sus colegas utilizaron NAMD para modelar la cápside del VIH, que se compone de más de 1, 300 proteínas idénticas que se ensamblan en una estructura similar a una caja que protege al virus hasta que ingresa a la célula huésped. Esa simulación representó las interacciones de más de 64 millones de átomos y requirió el uso de la supercomputadora Blue Waters en el Centro Nacional de Aplicaciones de Supercomputación de la U. de I. El nuevo estudio también hizo uso de Blue Waters, esta vez para mejorar la resolución del microscopio computacional.

Desde la izquierda, estudiante de posgrado Marcelo Melo, la profesora de química Zaida Luthey-Schulten, El investigador postdoctoral Rafael Bernardi y sus colegas han desarrollado un nuevo enfoque para modelar grandes interacciones moleculares a escalas atómicas y subatómicas. Su trabajo simplifica el método para otros científicos y estudiantes. Crédito:L. Brian Stauffer
El software NAMD está diseñado para describir el comportamiento de átomos individuales. Pero los átomos individuales involucrados en interacciones y reacciones químicas específicas no siempre se comportan como sus contrapartes en otros lugares. Comprender cómo varían requiere una mirada más cercana a las fuerzas subatómicas en juego. Esto es particularmente importante en las regiones dinámicas de moléculas, por ejemplo, aquellos lugares donde se forman o rompen enlaces químicos, dijeron los investigadores.
En el nuevo estudio, el equipo de investigación de Illinois se asoció con los expertos en QM Frank Neese, del Instituto Max Planck de Investigación del Carbón en Mulheim an der Ruhr, Alemania; y Gerd B. Rocha, de la Universidad Federal de Paraiba, en Joao Pessoa, Brasil.
Como demostración del nuevo enfoque, los investigadores simularon el comportamiento químico de los ARN de transferencia, moléculas que juegan un papel clave en la traducción de la información genética en proteínas. Usando NAMD, modelaron la estructura molecular general del tRNA en el momento en que una proteína especial carga un aminoácido en el tRNA. Dividieron dos sitios del complejo en regiones que requerían el enfoque mecánico cuántico más enfocado. (Vea una película de la simulación).
Las simulaciones subatómicas de las interacciones de las dos regiones permitieron al equipo ejecutar simulaciones de cuatro escenarios diferentes que permitirían que el tRNA funcionara como lo hace en la célula. Sus simulaciones revelaron que una de las cuatro vías químicas potenciales era más favorable energéticamente que las otras y, por lo tanto, era más probable que ocurriera.
Los investigadores también utilizaron varios métodos para dividir el complejo de ARNt entre las regiones MM y QM e informaron sobre cada enfoque.
"No elegimos solo una forma; elegimos tantos como fue posible. Le damos libertad al usuario. La forma en que lo estructura realmente depende del sistema particular que esté estudiando, "Dijo el investigador postdoctoral de la U. de I. Rafael Bernardi, coautor principal del estudio con el estudiante de posgrado Marcelo Melo.
"No hacemos todo el sistema de forma mecánica cuántica porque eso llevaría una eternidad calcular, "Dijo Melo.
"NAMD fue diseñado, y esta era la visión de mi esposo, para tratar sistemas realmente grandes, ", Dijo Luthey-Schulten." Ahora podemos agregar la escala subatómica a eso, abriendo vastas nuevas posibilidades para la investigación ".